Generellitet
Begrepet retinitt pigmentosa (RP) identifiserer en gruppe genetiske sykdommer preget av progressiv retinal degenerasjon.

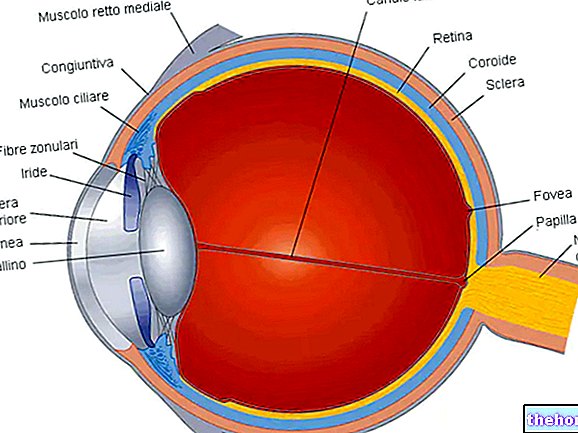
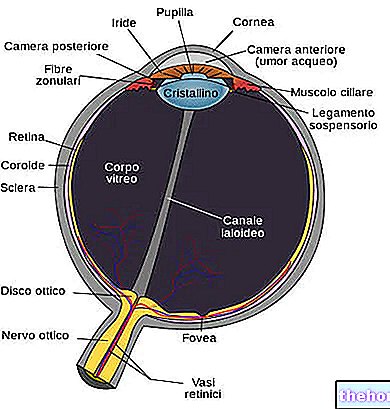
Retinitis pigmentosa er en retinal dystrofi preget av gradvis tap av fotoreseptorer og dysfunksjon av pigmentepitelet.Dette betyr at netthinnen gradvis reduserer evnen til å overføre visuell informasjon til hjernen via synsnerven.
Den patologiske prosessen begynner med endringer i retinalpigmentepitelet. Etter hvert som retinitis pigmentosa utvikler seg, blir det en tynning av blodårene som forsyner netthinnen, som gjennomgår atrofi. Ved undersøkelse av fundus er de karakteristiske avsetningene visuelt påviselige. Retinalpigment ( derav navnet fra sykdommen). Atrofiske endringer og skader kan også involvere synsnerven og gradvis dør de lysfølsomme cellene i netthinnen.
Pasienter som er rammet av retinitis pigmentosa opplever i utgangspunktet synsproblemer, spesielt i dårlig opplyste omgivelser, og klager over en innsnevring av det perifere synsfeltet. Sentral syn blir spart til de senere stadiene av sykdommen, og det endelige resultatet kan variere dramatisk: mange mennesker med retinitis pigmentosa beholder begrenset syn gjennom livet, mens andre mister synet helt.
Retinitis pigmentosa er en arvelig sykdom, hovedsakelig forårsaket av genetiske endringer som videreføres fra en eller begge foreldrene. Typen av genetisk defekt bestemmer hvilke netthinneceller som er mest involvert i lidelsen, og gjør det mulig å skille fra et klinisk synspunkt de forskjellige tilstandene. Til dags dato har mer enn 50 forskjellige genetiske defekter som er involvert i retinitis pigmentosa blitt identifisert. Unormaliteter kan overføres fra foreldre til avkom gjennom ett av tre arvsmønstre: autosomal recessiv, autosomal dominant eller heterosomal recessiv (X-bundet eller X-bundet).
Symptomer
For ytterligere informasjon: Retinitis Pigmentosa Symptomer
Retinitis pigmentosa finnes vanligvis hos ungdom og unge voksne. Symptomer vises ofte mellom 10 og 30 år, men diagnosen kan stilles i tidlig barndom eller mye senere i livet.
Tidlige symptomer på retinitt pigmentosa kan omfatte:
- Vanskeligheter med å se om natten (nattblindhet) eller i dårlige lysforhold
- Langsom tilpasning fra syn i mørket til det i lyset, og omvendt;
- Innsnevring av synsfeltet og tap av perifert syn;
- Følsomhet for lys og gjenskinn.
Noen symptomer avhenger av hvilken type fotoreseptorer som er involvert. Stengene er ansvarlige for svart -hvitt syn, mens kjeglene lar deg skille farger.
I de fleste tilfeller av retinitis pigmentosa er stengene involvert først. Imidlertid kan kjegler i de raskt utviklende formene også påvirkes på et tidlig stadium.
Stenger er konsentrert i de ytre delene av netthinnen og aktiveres av svakt lys, slik at degenerasjonen påvirker perifert og nattesyn. Hvis kjegler er involvert, er det mulig å oppleve tap av fargeoppfatning og sentralt syn.
Overvekten av de involverte fotoreseptorene bestemmes av den spesielle defekten som er tilstede i pasientens genetiske sammensetning.
Ofte er det første symptomet på retinitt pigmentosa nattblindhet (eller nocthalopia). Noen opplever at de trenger mer og mer tid til å tilpasse seg lysforskjeller når de beveger seg fra et godt opplyst område til et mørkere. En typisk form for synstap induserer innsnevring av perifert syn (tunnel- eller teleskopvisjon); dette mønsteret kalles et ringskotom. Noen ganger kan dette fenomenet mangle i de tidlige stadiene, men det blir lagt merke til når personen ofte snubler over objekter eller er involvert i en trafikkulykke. opplever vanskeligheter med å lese og detaljert arbeid som krever konsentrasjon på et enkelt objekt, for eksempel å tre en tråd gjennom et nåløye Mange pasienter rapporterer å se lysglimt (fotopsi), ofte beskrevet som små, flimrende og blinkende lys.
Sykdomsutviklingen og graden av synstap varierer fra person til person. Noen ekstreme tilfeller kan utvikle seg raskt i løpet av to tiår, andre et sakte forløp som aldri fører til fullstendig blindhet. Tidlig begynnelse finnes i mer alvorlige former for retinitis pigmentosa, mens pasienter med mildere tilstander (f.eks. Autosomaldominant) kan utvikle sykdommen i sitt femte eller sjette tiår av livet.I familier med X-linked retinitis pigmentosa blir menn rammet oftere enn kvinner og mer alvorlig; kvinner, derimot, overfører den genetiske egenskapen (de bærer det endrede genet på X -kromosomet) og viser symptomer på lidelsen sjeldnere.
Komplikasjoner
Retinitis pigmentosa vil fortsette å utvikle seg, om enn sakte. Fullstendig blindhet er imidlertid sjelden, men signifikant reduksjon i perifert og sentralt syn kan forekomme.
Pasienter med retinitt pigmentosa utvikler ofte hevelse i netthinnen (makulaødem) eller grå stær i en tidlig alder. Disse komplikasjonene kan behandles hvis de forstyrrer synet.
Relaterte sykdommer
Vanligvis har en pasient med retinitt pigmentosa ingen andre lidelser, og i dette tilfellet snakker vi om "ikke-syndromisk" eller enkel retinitt pigmentosa. Imidlertid deler flere syndromer noen kliniske symptomer med denne øyesykdommen; det vanligste er Usher syndrom, som rammer omtrent 10-30% av alle pasienter med retinitis pigmentosa og er forbundet med samtidig medfødt eller progressivt hørselstap. I Lebers medfødte amaurose kan imidlertid barn bli blinde, eller nesten blinde, i løpet av de første seks månedene av livet. Andre sykdommer knyttet til retinitis pigmentosa inkluderer Bardet-Biedl syndrom og Refsums sykdom.
Årsaker
Sykdommen kan være forårsaket av en rekke genetiske defekter: Faktisk er det flere gener som, hvis de påvirkes av endringen, kan forårsake retinitis pigmentosa fenotype. Disse koder normalt for proteiner involvert i transduksjonskaskaden som tillater syn, faktorer celletranskripsjon (som sender feilmeldinger til netthinneceller) eller for elementer som utgjør strukturen til fotoreseptorer. Arvelige genmutasjoner er tilstede i celler fra befruktningstidspunktet; vanlige abnormiteter inkluderer de hos RP1-gener (ved retinitt pigmentosa-1, autosomal dominant) , RHO (RP4, autosomal dominant) og RDS (RP7, autosomal dominant). Ikke-arvelige årsaker til retinitis pigmentosa er sjeldne, men muligheten for å finne et isolert tilfelle (spontan mutasjon), der det ikke er en familiehistorie av sykdommen.