Generellitet
Mitokondrielt DNA, eller mtDNA, er deoksyribonukleinsyren som ligger inne i mitokondriene, det vil si organeller i eukaryote celler som er ansvarlige for den svært viktige cellulære prosessen med oksidativ fosforylering.

Imidlertid har den også noen særegenheter, både strukturelle og funksjonelle, som gjør den unik i sitt slag. Disse særegenhetene inkluderer: sirkulæriteten til dobbeltstrengen av nukleotider, innholdet av gener (som bare er 37 elementer) og det nesten totale fraværet av ikke-kodende nukleotidsekvenser.
Mitokondrielt DNA utfører en grunnleggende funksjon for overlevelse av celler: det produserer enzymer som er nødvendige for å realisere oksidativ fosforylering.
Hva er mitokondrielt DNA?
Mitokondrielt DNA, eller mtDNA, er DNA plassert i mitokondriene.
Mitokondrier er de store cellulære organellene, typiske for eukaryote organismer, som omdanner kjemisk energi i maten til ATP, som er en energiform som kan utnyttes av celler.
BAKGRUNN PÅ STRUKTUR OG FUNKSJON AV MITOKONDRONER
Mitokondrier er rørformede, trådformede eller granulære i cytoplasma og opptar nesten 25% av volumet til sistnevnte.
De har to fosfolipid to -lags membraner, en ytterligere og en mer intern.
Den ytterste membranen, kjent som den ytre mitokondriemembranen, representerer omkretsen til hver mitokondrion og har transportproteiner (poriner og mer), som gjør den permeabel for molekyler med en størrelse lik eller mindre enn 5000 dalton.
Den innerste membranen, kjent som den indre mitokondriemembranen, inneholder alle de enzymatiske (eller enzymatiske) og koenzymkomponentene som er nødvendige for syntesen av ATP, og definerer et sentralt rom, kalt matrisen.
I motsetning til den ytterste membranen har den indre mitokondrielle membranen mange invaginasjoner - de såkalte ryggene - som øker det totale arealet.
Mellom de to mitokondrielle membranene er det et mellomrom på nesten 60-80 Ångstrøm (A). Dette rommet kalles intermembranrommet. Intermembranrommet har en sammensetning som er veldig lik cytoplasma.
Syntesen av ATP, som drives av mitokondriene, er en veldig kompleks prosess, som biologer identifiserer med begrepet oksidativ fosforylering.
NØYAKTIG BELIGGENHET FOR MITOKONDRALT DNA OG KVANTITET

Figur: en menneskelig mitokondrion.
Mitokondrielt DNA ligger i den mitokondrielle matrisen, dvs. i rommet avgrenset av den indre mitokondriemembranen.
Ifølge pålitelige vitenskapelige studier kan hver mitokondrion inneholde fra 2 til 12 kopier av mitokondrielt DNA.
Gitt det faktum at noen celler i menneskekroppen kan inneholde flere tusen mitokondrier i dem, kan det totale antallet kopier av mitokondrielt DNA i en enkelt menneskelig celle nå opptil 20 000 enheter.
Vær oppmerksom på: Antallet mitokondrier i humane celler varierer avhengig av celletype. For eksempel kan hepatocytter (dvs. leverceller) inneholde mellom 1000 og 2000 mitokondrier hver, mens erytrocytter (dvs. røde blodlegemer) er totalt blottet for dem.
Struktur
Den generelle strukturen til et mitokondrielt DNA -molekyl ligner den generelle strukturen til kjernefysisk DNA, det vil si den genetiske arven som er tilstede i kjernen til eukaryote celler.
Faktisk, analogt med kjernefysisk DNA:
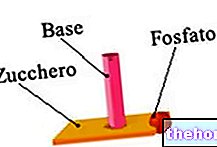
- Mitokondrielt DNA er en biopolymer, som består av to lange tråder av nukleotider. Nukleotider er organiske molekyler som stammer fra forening av tre elementer: et sukker med 5 karbonatomer (for DNA, deoksyribose), en nitrogenbasert base og en fosfatgruppe.
- Hvert nukleotid i mitokondrielt DNA binder seg til neste nukleotid i samme streng ved hjelp av en fosfodiesterbinding mellom karbon 3 i dets deoksyribose og fosfatgruppen til det umiddelbart følgende nukleotidet.
- De to strengene av mitokondrielt DNA har motsatt orientering, med enden av den ene i samspill med enden av den andre og omvendt. Dette bestemte arrangementet er kjent som det antiparallelle arrangementet (eller antiparallell orientering).
- De to trådene av mitokondrielt DNA samhandler med hverandre gjennom de nitrogenholdige basene.
Nærmere bestemt etablerer hver nitrogenbase i hver filament hydrogenbindinger med en og bare en nitrogenbasert base, til stede på den andre filamenten.
Denne typen interaksjon kalles "sammenkobling mellom nitrogenholdige baser" eller "par nitrogenholdige baser". - De nitrogenholdige basene i mitokondrielt DNA er adenin, tymin, cytosin og guanin.
Paringen som disse nitrogenholdige basene gir opphav til, er ikke tilfeldig, men svært spesifikk: adenin interagerer bare med tymin, mens cytosin bare interagerer med guanin. - Mitokondrielt DNA er hjemsted for gener (eller gensekvenser). Gener er sekvenser med mer eller mindre lange nukleotider, med en veldefinert biologisk betydning. I de fleste tilfeller gir de opphav til proteiner.

STRUKTURELLE BETINGELSER OM MITOKONDRAL DNA
Utover de nevnte analogiene har menneskelig mitokondrielt DNA noen strukturelle særegenheter, som skiller det betydelig fra menneskelig atom -DNA.
For det første er det et sirkulært molekyl, mens kjernefysisk DNA er et lineært molekyl.
Dermed har den 16 569 nitrogenholdige basepar, mens kjernefysisk DNA har hele 3,3 milliarder.
Den inneholder 37 gener, mens kjernefysisk DNA ser ut til å inneholde mellom 20 000 og 25 000.
Det er ikke organisert i kromosomer, mens kjernefysisk DNA er delt inn i 23 kromosomer og danner, med noen spesifikke proteiner, et stoff som kalles kromatin.
Til slutt inkluderer den en rekke nukleotider som deltar i to gener samtidig, mens kjernefysisk DNA har gener hvis nukleotidsekvenser er veldefinerte og forskjellige fra hverandre.
Opprinnelse
Mitokondrielt DNA har mest sannsynlig en "bakteriell" opprinnelse.
Faktisk, på grunnlag av mange uavhengige studier, tror molekylærbiologer at den cellulære tilstedeværelsen av mitokondrielt DNA er et resultat av inkorporering av forfedre eukaryote celler av uavhengige bakterieorganismer, veldig lik mitokondrier.
Denne merkelige oppdagelsen har bare delvis overrasket det vitenskapelige samfunnet, ettersom DNA som er tilstede i bakterier generelt er en sirkulær nukleotidstreng, som mitokondrielt DNA.
Teorien om hvilken mitokondrier og mitokondrielt DNA har en "bakteriell opprinnelse, tar navnet" endosymbiotisk teori ", fra ordet" endosymbiose. "Kort sagt, i biologien, indikerer begrepet" endosymbiose "et samarbeid mellom to organismer, som involverer "inkorporering av det ene i det andre for å oppnå en viss fordel.
Nysgjerrighet
Ifølge pålitelige vitenskapelige studier ville mange bakteriegener i evolusjonsforløpet, som er tilstede på det fremtidige mitokondrielle DNA, ha endret plassering og flyttet inn i kjernefysisk DNA.
Med andre ord, i begynnelsen av endosymbiose, befant noen gener som nå er tilstede på kjernefysisk DNA i DNA -en til de bakterielle organismer, som senere skulle bli mitokondrier.
For å støtte teorien om et skift av gener mellom mitokondrielt DNA og kjernefysisk DNA, er observasjonen at visse gener stammer fra mitokondrielt DNA, hos noen arter og fra kjernefysisk DNA, i andre.
Funksjon
Mitokondrielt DNA produserer enzymer (dvs. proteiner), nødvendige for korrekt implementering av den delikate oksidative fosforyleringsprosessen.
Instruksjonene for syntetisering av disse enzymene ligger i de 37 genene som utgjør mitokondrielt DNA -genom.
HVILKE MITOKONDRALE DNA -GENER KODE: DETALJENE
De 37 genene i mitokondriell DNA -kode for: proteiner, tRNA og rRNA.
Spesielt:
- 13 koder for 13 proteiner som er ansvarlige for å utføre oksidativ fosforylering
- 22 kode for 22 tRNA -molekyler
- 2 koder for 2 rRNA -molekyler
Molekylene til tRNA og rRNA er grunnleggende for syntesen av de nevnte 13 proteinene, ettersom de utgjør maskinen som regulerer produksjonen.
Så, med andre ord, mitokondrielt DNA besitter informasjonen for å produsere et bestemt sett med proteiner og verktøyene som er nødvendige for syntesen.
Hva er RNA, tRNA og rRNA?
RNA, eller ribonukleinsyre, er nukleinsyren som spiller en grunnleggende rolle i dannelsen av proteiner, med utgangspunkt i DNA.
Vanligvis enkelstrenget, kan ANN eksistere i forskjellige former (eller typer), avhengig av den spesifikke funksjonen den er delegert til.
TRNA og rRNA er to av disse mulige formene.
TRNA brukes til å tilsette aminosyrer under prosessen med å lage proteiner. Aminosyrer er de molekylære enhetene som utgjør proteiner.
RRNA danner ribosomene, det vil si mobilstrukturene der syntesen av proteiner finner sted.
For å vite detaljert ANN og funksjonene, kan leserne klikke her.
FUNKSJONelle opplysninger om mitokondralt DNA
Fra et funksjonelt synspunkt har mitokondrielt DNA noen særegne egenskaper som tydelig skiller det fra kjernefysisk DNA.
Her er hva disse særegne egenskapene består av:
- Mitokondrielt DNA er semi-uavhengig, i den forstand at det trenger inngrep fra noen proteiner syntetisert fra kjernefysisk DNA.
På den annen side er kjernefysisk DNA helt autonomt og produserer i seg selv alt det trenger for å utføre oppgavene på riktig måte. - Mitokondrielt DNA har en litt annen genetisk kode enn kjernefysisk DNA. Dette fører til en rekke forskjeller i fremstilling av proteiner: hvis en bestemt sekvens av nukleotider i kjernefysisk DNA fører til dannelsen av et bestemt protein, fører den samme sekvensen i mitokondrielt DNA til dannelsen av et litt annet protein.
- Mitokondrielt DNA har svært få ikke-kodende nukleotidsekvenser, det vil si at de ikke produserer proteiner, tRNA eller rRNA. I prosent er bare 3% av mitokondrielt DNA ikke-kodende.
På den annen side er kjernefysisk DNA bare 7% kodende, så det inneholder mange ikke-kodende nukleotidsekvenser (hele 93%).
Tabell: sammendrag av forskjellene mellom humant mitokondrielt DNA og humant atom -DNA.
Mitokondrielt DNA
Kjernefysisk DNA
- Det er sirkulært
- Det er lineært
- Den har 16 569 nitrogenholdige basepar i alt
- Den har totalt 3,3 milliarder nitrogenholdige basepar
- Den inneholder 37 gener i alt
- Den inneholder mellom 20.000 og 25.000 gener
- For å fungere skikkelig trenger den støtte fra noen genprodukter, som stammer fra kjernefysisk DNA
- Den er autonom og produserer i seg selv alt den trenger for å utføre sine funksjoner
- Den kan finnes i flere kopier innenfor hver enkelt mitokondrion
- Det er unikt, det vil si at det bare er i ett eksemplar, og det ligger i kjernen
- 97% av nukleotidsekvensen som komponerer den, koder
- Bare 7% av nukleotidsekvensen som komponerer den, koder
- Det er ikke organisert i kromosomer
- Det er delt inn i 23 kromosomer
- Den bruker en genetisk kode som er litt annerledes enn den så å si "tradisjonelle"
- Bruk den "tradisjonelle" genetiske koden
- Arven er mors
- Arven er halvparten av moren og halvparten av faren
- Noen av dets nukleotider deltar i to gener samtidig
- Sekvensene av nukleotider som utgjør genene skiller seg godt fra hverandre
Arv
Mitokondriell DNA -arv er strengt mors.
Dette betyr at i et foreldrepar er det kvinnen som overfører mitokondrielt DNA til avkommet (dvs. til barna).
På en helt motsatt måte til det ovennevnte, er arvelig kjerne -DNA halvparten mors og halvt fedre, med andre ord, begge foreldrene bidrar likt til overføring av kjernefysisk DNA i avkom.
Vær oppmerksom på: mors arv av mitokondrielt DNA involverer også mitokondriell struktur. Derfor er mitokondriene tilstede i et individ mors.
Tilknyttede patologier
Premiss: En genetisk mutasjon er en permanent endring i sekvensen av nukleotider, som utgjør et kjernefysisk eller mitokondrielt DNA -gen.
Vanligvis resulterer tilstedeværelsen av en genetisk mutasjon i en "endring eller tap av normal funksjon av det involverte genet.
Tilstedeværelsen av mutasjoner i mitokondrielle DNA -gener kan føre til et bredt spekter av sykdommer, inkludert:
- Lebers arvelige optiske nevropati
- Kearns-Sayre syndrom
- Leighs syndrom
- Mangel på cytokrom C -oksidase
- Progressiv ekstern oftalmoplegi
- Pearsons syndrom
- Mitokondriell encefalomyopati med melkesyreacidose og slaglignende episoder (MELAS syndrom)
- Diabetes med maternelt overført døvhet
- Myoklonisk epilepsi med uregelmessige røde fibre
Når det gjelder de patologiske forholdene knyttet til en eller flere mitokondrielle DNA -mutasjoner, må to aspekter avklares.
For det første avhenger sykdommens alvorlighetsgrad av det kvantitative forholdet mellom muterte mitokondrielle DNA og friske, normale mitokondrielle DNA. Hvis antallet muterte mitokondrielle DNA er langt større enn for friske DNA, vil den resulterende tilstanden bli mer alvorlig.
For det andre påvirker mutasjoner i mitokondrielt DNA bare noen vev i organismen, spesielt de som krever store mengder ATP som følge av den oksidative fosforyleringsprosessen. Dette er ganske forståelig: for å lide mer enn én funksjonsfeil i mitokondrielt DNA er cellene som trenger mest funksjonen som mitokondrielt DNA normalt oppfyller.
LEBER ERFARLIG OPTISK NEUROPATI
Lebers arvelige optiske nevropati oppstår som et resultat av mutasjon av så mange som fire mitokondrielle DNA -gener. Disse genene inneholder informasjonen som fører til syntesen av den såkalte kompleks I (eller NADH oksidreduktase), et av de forskjellige enzymene som er involvert i den oksidative fosforyleringsprosessen.
Manifestasjonene av patologien består i en progressiv degenerasjon av synsnerven og et gradvis synstap.
KEARNS-SAYRE SYNDROME
Kearns-Sayre syndrom vises på grunn av mangel på en god del mitokondrielt DNA (N.B: mangelen på en bestemt nukleotidsekvens kalles en sletting).
Personer med Kearns-Sayre syndrom utvikler oftalmoplegi (total eller delvis lammelse av okulomotoriske muskler), en form for retinopati og abnormiteter i hjerterytmen (atrioventrikulær blokk).
LEIGHS SYNDROME
Leigh syndrom oppstår som et resultat av mitokondrielle DNA-mutasjoner, som kan påvirke ATP-syntaseproteinet (også kalt V-kompleks) og / eller noen tRNA.
Leighs syndrom er en progressiv nevrologisk sykdom som oppstår i barndommen eller i barndommen og er ansvarlig for: forsinket utvikling, muskelsvakhet, perifer nevropati, motoriske lidelser, pustevansker og oftalmoplegi.
Mangel på CYTOCHROME C OXIDASE
Cytokrom C -oksidasemangel oppstår på grunn av mutasjon av minst 3 mitokondrielle DNA -gener. Disse genene er avgjørende for riktig syntese av cytokrom C -oksidase (eller komplekst IV) enzym, involvert i den oksidative fosforyleringsprosessen.
Typiske manifestasjoner av cytokrom C -oksidasemangel består av: skjelettmuskeldysfunksjon, hjertedysfunksjon, nedsatt nyrefunksjon og nedsatt leverfunksjon.
PROGRESSIV EKSTERN OPTALMOPLEGI
Progressiv ekstern oftalmoplegi oppstår fra mangel på et betydelig antall mitokondrielle DNA -nukleotider (sletting)
Med en progressiv karakter (som man kan gjette fra navnet), forårsaker denne patologien en lammelse av de okulomotoriske musklene, med påfølgende ptose og betydelige synsproblemer.
PEARSONS SYNDROME
Pearsons syndrom vises etter en iøynefallende sletting av mitokondrielt DNA, på lignende måte som progressiv ekstern oftalmoplegi og Kearns-Sayre syndrom.
Typiske manifestasjoner av Pearsons syndrom består av: sideroblastisk anemi, bukspyttkjerteldysfunksjon (f.eks. Insulinavhengig diabetes), nevrologiske underskudd og muskelsykdommer.
Pearson syndrom får vanligvis den berørte personen til å dø i ung alder. Faktisk når de som er berørt av denne patologien sjelden voksen alder.
MELAS SYNDROME
MELAS syndrom, også kjent som mitokondriell encefalomyopati med melkesyreacidose og slaglignende episoder, oppstår fra mutasjon av minst 5 mitokondrielle DNA-gener.
Disse genene bidrar til syntesen av NADH-oksid-reduktase, eller kompleks I, og av noen tRNA-er.
MELAS syndrom innebærer tilstedeværelse av nevrologiske lidelser, muskelsykdommer, uvanlig opphopning av melkesyre i vevet (med alle symptomene som følger med), pusteproblemer, tap av tarmfunksjonskontroll, tilbakevendende tretthet, nyreproblemer, hjerteproblemer, diabetes, epilepsi og mangel på koordinering.
ANDRE PATOLOGIER
Ifølge forskjellige vitenskapelige studier vil sykdommer som syklisk oppkastssyndrom, retinitis pigmentosa, ataksi, Parkinsons sykdom og Alzheimers sykdom også se involvering av mitokondrielt DNA og noen av dets mutasjoner.