Aktive ingredienser: Prucalopride
Resolor 1 mg filmdrasjerte tabletter
Resolor 2 mg filmdrasjerte tabletter
Indikasjoner Hvorfor brukes Resolor? Hva er den til?
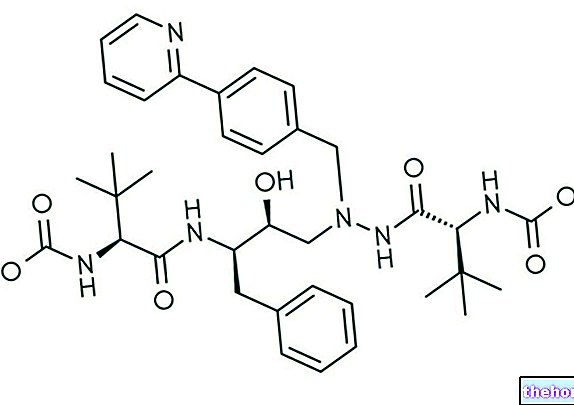
Resolor inneholder virkestoffet prukaloprid.
Resolor tilhører en gruppe medisiner som forbedrer tarmmotiliteten (gastrointestinal prokinetikk). Den virker på tarmens muskelvegg og hjelper til med å gjenopprette normal funksjon. Resolor brukes til å behandle kronisk forstoppelse hos voksne der avføringsmidler ikke fungerer som de skal.
Skal ikke brukes til barn og ungdom under 18 år.
Kontraindikasjoner Når Resolor ikke skal brukes
Ikke ta Resolor
- hvis du er allergisk mot prukaloprid eller noen av de andre innholdsstoffene i dette legemidlet (listet opp i avsnitt 6),
- hvis du gjennomgår nyredialyse,
- hvis du har perforering eller obstruksjon av tarmveggen, alvorlig betennelse i tarmkanalen, som Crohns sykdom, ulcerøs kolitt eller giftig megakolon / megarekt.
Forholdsregler for bruk Hva du trenger å vite før du bruker Resolor
Snakk med legen din før du tar Resolor.
Vær spesielt forsiktig med Resolor og fortell legen din:
- hvis du har alvorlig nyresykdom,
- hvis du har alvorlig leversykdom,
- hvis du for øyeblikket er under medisinsk tilsyn for et alvorlig helseproblem, for eksempel en hjerte- eller lungesykdom, en psykisk eller nervøs lidelse, kreft, AIDS eller en hormonell lidelse.
Interaksjoner Hvilke medisiner eller matvarer kan endre effekten av Resolor
Fortell legen din dersom du bruker, nylig har brukt eller planlegger å bruke andre legemidler.
Resolor med mat og drikke
Resolor kan tas med eller uten mat og drikke når som helst på dagen.
Advarsler Det er viktig å vite at:
Graviditet og amming
Resolor anbefales ikke under graviditet.
- Fortell legen din dersom du er gravid eller planlegger å bli gravid.
- Bruk en pålitelig prevensjonsmetode under behandling med Resolor for å unngå å bli gravid.
- Informer legen din dersom du blir gravid mens du bruker Resolor.
Under amming kan prukaloprid gå over i morsmelk. Amming anbefales ikke under behandling med Resolor. Rådfør deg med legen din om dette.
Spør legen din om råd før du tar medisiner.
Kjøring og bruk av maskiner
Resolor påvirker neppe din evne til å kjøre bil eller bruke maskiner. Imidlertid kan Resolor i noen tilfeller forårsake svimmelhet og tretthet, spesielt den første behandlingsdagen, og dette kan påvirke bilkjøring og bruk av maskiner.
Resolor inneholder laktose
Hvis legen din har fortalt deg at du ikke tåler noen sukkerarter, må du kontakte legen din før du tar dette legemidlet.
Dose, metode og administrasjonstidspunkt Hvordan bruke Resolor: Dosering
Ta alltid dette legemidlet nøyaktig som beskrevet i dette pakningsvedlegget eller som foreskrevet av legen din.
Rådfør deg med lege eller apotek hvis du er i tvil. Fortsett å ta Resolor hver dag i den tiden legen din har angitt. Legen din vil kanskje vurdere tilstanden din og fordelen med langvarig behandling på nytt etter de første 4 ukene og deretter med jevne mellomrom.
Den vanlige dosen Resolor for de fleste pasienter er en 2 mg tablett en gang daglig.
Hvis du er over 65 år eller har alvorlig leversykdom, er startdosen en 1 mg tablett en gang daglig; om nødvendig kan legen øke dosen til 2 mg en gang daglig.
Legen din kan også anbefale den lavere dosen på en 1 mg tablett en gang daglig hvis du har alvorlig nyresykdom.
Å ta mer enn anbefalt dose vil ikke øke medisinens effektivitet.
Resolor er kun indisert for voksne og bør ikke tas av barn eller ungdom opp til 18 år.
Overdosering Hva du skal gjøre hvis du har tatt for mye Resolor
Dersom du tar for mye av Resolor
Det er viktig å holde seg til dosen som legen din foreskriver. Hvis du har tatt mer Resolor enn foreskrevet, kan du oppleve diaré, hodepine og / eller kvalme. Ved diaré må du drikke nok vann.
Dersom du har glemt å ta Resolor
Ikke ta en dobbel dose som erstatning for en glemt tablett. Ta din neste dose til vanlig tid.
Hvis du slutter å bruke Resolor
Hvis du stopper Resolor, kan symptomene på forstoppelse komme tilbake.
Spør lege eller apotek hvis du har ytterligere spørsmål om bruken av dette legemidlet
Bivirkninger Hva er bivirkningene av Resolor
Som alle andre legemidler kan dette legemidlet forårsake bivirkninger, men ikke alle får det. Uønskede effekter oppstår hovedsakelig ved behandlingsstart og forsvinner vanligvis innen få dager etter fortsatt behandling.
Følgende bivirkninger har forekommet svært ofte (kan ramme flere enn 1 av 10 personer): hodepine, kvalme, diaré og magesmerter.
Følgende bivirkninger har ofte oppstått (kan forekomme hos opptil 1 av 10 personer): redusert appetitt, svimmelhet, oppkast, fordøyelsesforstyrrelser (dyspepsi), flatulens, unormal tarmborborygma, tretthet.
Følgende uvanlige bivirkninger (kan forekomme hos opptil 1 av 100 personer) er også rapportert: skjelvinger, hjertebank, rektal blødning, økt vannlating (pollakiuri), feber og kvalme. Fortell legen din dersom hjertebank oppstår.
Rapportering av bivirkninger
Rådfør deg med lege eller apotek dersom du får bivirkninger, inkludert mulige bivirkninger som ikke er nevnt i dette pakningsvedlegget. Du kan også rapportere bivirkninger direkte via det nasjonale rapporteringssystemet som er oppført i vedlegg V. Ved å rapportere bivirkninger kan du hjelpe til med å gi mer informasjon om sikkerheten til dette legemidlet.
Utløp og oppbevaring
Hold denne medisinen utilgjengelig for barn.
Ikke bruk dette legemidlet etter utløpsdatoen som er angitt på blisteren og esken etter Utløpsdatoen refererer til den siste dagen i den måneden.
Oppbevares i den originale blisterpakningen for å beskytte mot fuktighet.
Ikke kast medisiner i avløpsvann eller husholdningsavfall. Spør apoteket om hvordan du skal kaste medisiner du ikke bruker lenger. Dette vil bidra til å beskytte miljøet.
Hva Resolor inneholder
Den aktive ingrediensen er prukaloprid.
En Resolor 1 mg filmdrasjert tablett inneholder 1 mg prukaloprid (som suksinat).
En Resolor 2 mg filmdrasjert tablett inneholder 2 mg prukaloprid (som suksinat).
Andre innholdsstoffer er:
Laktosemonohydrat (se avsnitt 2), mikrokrystallinsk cellulose, kolloidalt kiseldioksid, magnesiumstearat, hypromellose, triacetin, titandioksid (E171), makrogol. 2 mg tabletten inneholder også rødt jernoksid (E172), gult jernoksid (E172), indigokarmin -innsjø (E132).
Beskrivelse av hvordan Resolor ser ut og innholdet i pakningen
Resolor 1 mg filmdrasjerte tabletter er hvite til off-white, runde tabletter merket "PRU 1" på den ene siden.
Resolor 2 mg filmdrasjerte tabletter er rosa, runde tabletter med "PRU 2" preget på den ene siden.
Resolor er tilgjengelig i aluminium / aluminium perforert enhetsdoseblister (kalender) som inneholder 7 tabletter. Hver pakning inneholder 7x1, 14x1, 28x1 eller 84x1 filmdrasjerte tabletter.
Det er ikke sikkert at alle pakningsstørrelser blir markedsført.
Kildepakningsvedlegg: AIFA (Italian Medicines Agency). Innhold publisert i januar 2016. Informasjonen som er tilstede er kanskje ikke oppdatert.
For å få tilgang til den mest oppdaterte versjonen, er det lurt å gå til nettstedet til AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttig informasjon.
01.0 LEGEMIDLETS NAVN
RESOLOR 1 MG TABLETTER DREKKET MED FILM
02.0 KVALITATIV OG KVANTITATIV SAMMENSETNING
Hver filmdrasjerte tablett inneholder 1 mg prukaloprid (som suksinat).
Hjelpestoffer med kjent effekt: Hver filmdrasjerte tablett inneholder 142,5 mg laktose (som monohydrat).
For fullstendig liste over hjelpestoffer, se pkt.6.1.
03.0 LEGEMIDDELFORM
Filmdrasjert tablett (tablett).
Hvite til off-white, runde, bikonvekse tabletter, preget med "PRU 1" på den ene siden.
04.0 KLINISK INFORMASJON
04.1 Terapeutiske indikasjoner
Resolor er indisert for symptomatisk behandling av kronisk forstoppelse hos voksne som avføringsmidler ikke gir tilstrekkelig lindring for.
04.2 Dosering og administrasjonsmåte
Dosering
Voksne: 2 mg én gang daglig med eller uten mat, når som helst på dagen.
På grunn av den spesifikke virkningsmåten til prukaloprid (stimulering av fremdriftsmotilitet) forventes det ikke at en daglig dose større enn 2 mg vil føre til økt effekt.
Hvis prukaloprid en gang daglig ikke er effektivt etter 4 ukers behandling, må pasienten undersøkes på nytt og nytten av å fortsette behandlingen bør vurderes.
Effekten av prukaloprid er påvist i dobbeltblinde placebokontrollerte studier med en varighet på opptil tre måneder.Effekt utover tre måneder er ikke påvist i placebokontrollerte studier (se pkt.5.1). Ved langvarig behandling bør fordelen revurderes med jevne mellomrom.
Spesielle populasjoner
Eldre (> 65 år): Start med 1 mg en gang daglig (se pkt. 5.2); om nødvendig kan dosen økes til 2 mg en gang daglig.
Pasienter med nyreinsuffisiens: Dosen for pasienter med alvorlig nedsatt nyrefunksjon (GFR 2) er 1 mg én gang daglig (se pkt. 4.3 og 5.2). Ingen dosejustering er nødvendig for pasienter med mild til moderat nedsatt nyrefunksjon.
Pasienter som lider av leverinsuffisiens: Pasienter med alvorlig nedsatt leverfunksjon (Child-Pugh klasse C) starter med en dose på 1 mg én gang daglig, som kan økes til 2 mg om nødvendig for å forbedre effekten og hvis dosen på 1 mg tolereres godt (se pkt.4.4 og 5.2 ) Ingen dosejustering er nødvendig for pasienter med mild til moderat nedsatt leverfunksjon.
Pediatrisk populasjon: Resolor skal ikke brukes til barn og ungdom under 18 år (se pkt.5.1).
Administrasjonsmåte
Oral bruk.
04.3 Kontraindikasjoner
• Overfølsomhet overfor virkestoffet eller overfor noen av hjelpestoffene listet opp i pkt. 6.1.
• Nyresvikt som krever dialyse.
• Tarmperforering eller obstruksjon på grunn av strukturelle eller funksjonelle forstyrrelser i tarmveggen, obstruktiv ileus, alvorlige inflammatoriske tilstander i tarmkanalen som Crohns sykdom og ulcerøs kolitt, samt giftig megakolon / megarekt.
04.4 Spesielle advarsler og passende forholdsregler for bruk
Den viktigste eliminasjonsmåten for prukaloprid er renal utskillelse (se pkt. 5.2). En dose på 1 mg anbefales til personer med alvorlig nedsatt nyrefunksjon (se pkt. 4.2).
Resolor bør forskrives med forsiktighet til pasienter med alvorlig nedsatt leverfunksjon (Child-Pugh klasse C), fordi data om bruk hos slike pasienter er begrenset (se pkt.4.2).
Sikkerhet og effekt av Resolor til bruk hos pasienter med samtidig alvorlig og klinisk ustabil sykdom (for eksempel hjerte- eller lungesykdom, nevrologiske eller psykiatriske lidelser, kreft eller AIDS og andre endokrine lidelser) har ikke blitt fastslått i kontrollerte kliniske studier. Resolor bør forskrives med forsiktighet til pasienter med disse tilstandene, spesielt når det brukes til pasienter med arytmi eller iskemisk kardiovaskulær sykdom.
Ved alvorlig diaré kan effekten av orale prevensjonsmidler reduseres; Derfor anbefales bruk av en annen prevensjonsmetode for å forhindre mulig ineffektivitet av oral prevensjon (se informasjon om forskrivning av orale prevensjonsmidler).
Tablettene inneholder laktose. Pasienter med sjeldne arvelige problemer med galaktoseintoleranse, Lapp-laktasemangel eller glukose-galaktosemalabsorpsjon bør ikke ta denne medisinen.
04.5 Interaksjoner med andre legemidler og andre former for interaksjon
Prucaloprid har et lavt farmakokinetisk interaksjonspotensial. Det utskilles stort sett uendret i urinen (ca. 60% av dosen) og metabolismen in vitro det er veldig sakte.
Prucaloprid hemmet ikke spesifikke CYP450 -aktiviteter i studiene in vitro i humane levermikrosomer, ved terapeutisk relevante konsentrasjoner.
Selv om prukaloprid kan være et svakt substrat for P-glykoprotein (P-gp), er det ikke en P-gp-hemmer ved klinisk relevante konsentrasjoner.
Effekter av prukaloprid på farmakokinetikken til andre legemidler
Det var en 30% økning i plasmakonsentrasjoner av erytromycin under samtidig behandling med prukaloprid. Mekanismen bak denne interaksjonen er uklar.
Prucaloprid hadde ingen klinisk relevante effekter på farmakokinetikken til warfarin, digoksin, alkohol og paroksetin eller orale prevensjonsmidler.
Effekter av andre legemidler på farmakokinetikken til prukaloprid
Ketokonazol (200 mg to ganger daglig), en kraftig hemmer av CYP3A4 og P-gp, økte systemisk eksponering for prukaloprid med omtrent 40%. Denne effekten er for liten til å være klinisk relevant. Interaksjoner av lignende betydning kan forventes med andre potente P -gp -hemmere som verapamil, cyklosporin A og kinidin.
Terapeutiske doser av probenecid, cimetidin, erytromycin og paroksetin påvirket ikke farmakokinetikken til prukaloprid.
04.6 Graviditet og amming
Kvinner i fertil alder
Kvinner i fertil alder bør bruke effektive prevensjonsmetoder under behandling med prukaloprid.
Svangerskap
Erfaring med prukaloprid i svangerskapet er begrenset. Tilfeller av spontan abort har blitt observert i kliniske studier, men i nærvær av andre risikofaktorer er forholdet til prukaloprid ukjent. Dyrestudier indikerer ikke direkte eller skadelige effekter. Indirekte effekter på graviditet, embryonal / fosterutvikling, fødsel eller postnatal utvikling (se pkt. 5.3) Resolor anbefales ikke under graviditet.
Foringstid
Prucaloprid skilles ut i morsmelk. Ved terapeutiske doser av Resolor forventes det imidlertid ingen effekt på nyfødte / spedbarn som ammes. I mangel av menneskelige data, anbefales ikke bruk av Resolor under amming.
Fruktbarhet
Dyrestudier indikerer at det ikke er noen effekt på fruktbarhet hos menn eller kvinner.
04.7 Påvirkning av evnen til å kjøre bil og bruke maskiner
Resolor kan svekke evnen til å kjøre bil og bruke maskiner noe, ettersom svimmelhet og tretthet ble observert i kliniske studier, spesielt den første behandlingsdagen (se pkt. 4.8).
04.8 Bivirkninger
Oppsummering av sikkerhetsprofilen
Det ble utført en integrert analyse av 17 dobbeltblinde placebokontrollerte studier der Resolor ble administrert oralt til ca. 3300 pasienter med kronisk forstoppelse. Av disse pasientene tok over 1500 Resolor i anbefalt dose på 2 mg per dag, mens ca. 1360 var behandlet med 4 mg prukaloprid per dag. De hyppigst rapporterte bivirkningene forbundet med Resolor 2 mg -behandling er hodepine (17,8%) og gastrointestinale symptomer (magesmerter (13,7%), kvalme (13,7%) og diaré (12,0%)). Bivirkninger oppstår hovedsakelig ved behandlingsstart og forsvinner vanligvis innen få dager etter fortsatt behandling. Andre bivirkninger har blitt rapportert av og til. De fleste bivirkningene var "milde til moderate" i intensitet.
Tabell over bivirkninger
Følgende bivirkninger ble rapportert i kontrollerte kliniske studier med anbefalt dose på 2 mg med frekvenser som tilsvarer: svært vanlige (≥ 1/10), vanlige (≥ 1/100,
Beskrivelse av noen bivirkninger
Etter den første behandlingsdagen ble de vanligste bivirkningene rapportert med lignende frekvens (forskjell i forekomst på ikke mer enn 1% mellom prukaloprid og placebo) under behandling med Resolor og under behandling med placebo, med unntak av kvalme og diaré som fortsatte å forekomme oftere under Resolor -terapi, men mindre uttalt (forskjeller i forekomst mellom Resolor og placebo på henholdsvis 1,3% (kvalme) og 3,4% (diaré)).
Hjertebank ble rapportert hos 0,7% av pasientene som ble behandlet med placebo, 0,9% av pasientene som ble behandlet med 1 mg prukaloprid, 0,9% av pasientene som ble behandlet med 2 mg prukaloprid og 1,9% av pasientene som ble behandlet med 4 mg prukaloprid. De fleste pasientene fortsatte å ta prukaloprid. Pasienter bør diskutere med den behandlende legen den nye starten på hjertebank, samt utseendet på nye symptomer.
Rapportering av mistenkte bivirkninger
Rapportering av mistenkte bivirkninger som oppstår etter godkjenning av legemidlet er viktig, da det muliggjør kontinuerlig overvåking av nytte / risiko -forholdet til legemidlet Helsepersonell bes rapportere alle mistenkte bivirkninger via Det italienske legemiddelkontoret. . Nettsted: http://www.agenziafarmaco.gov.it/it/responsabili.
04.9 Overdosering
I en studie med friske frivillige ble behandling med prukaloprid godt tolerert ved administrering i et eskalerende regime opp til 20 mg en gang daglig (10 ganger anbefalt terapeutisk dose). En overdose kan resultere i symptomer som "forsterker de kjente farmakodynamiske effektene av prukaloprid og inkluderer hodepine, kvalme og diaré. Ingen spesifikk behandling er tilgjengelig for Resolor -overdose. Skulle en overdose oppstå, bør pasienten behandles symptomatisk og bør behandles". Støttende tiltak som iverksettes etter behov Overdreven væsketap forårsaket av diaré eller oppkast kan kreve korrigering av elektrolyttubalanse.
05.0 FARMAKOLOGISKE EGENSKAPER
05.1 Farmakodynamiske egenskaper
Farmakoterapeutisk gruppe: andre avføringsmidler, ATC -kode: A06AX05.
Virkningsmekanismen
Prucalopride er et dihydrobenzofurankarboksamid med gastrointestinal prokinetisk aktivitet. Prucalopride er en selektiv serotonin (5-HT4) reseptoragonist med høy affinitet, som sannsynligvis forklarer dens prokinetiske effekter. In vitro en "affinitet for andre reseptorer ble bare funnet i konsentrasjoner som overstiger dets affinitet for 5-HT4-reseptoren med minst 150 ganger. Hos rotter, prukaloprid in vivo, ved doser over 5 mg / kg (30-70 ganger den kliniske eksponeringen og utover), induserer det hyperprolaktinemi forårsaket av en antagonistvirkning mot D2-reseptoren.
Hos hunder endrer prukaloprid tarmmotilitetsmønstre gjennom stimulering av serotonin 5-HT4-reseptoren: det stimulerer proksimal tarmmotilitet, forbedrer gastroduodenal motilitet og akselererer forsinket tømming av magesekken. Prucalopride induserer også gigantiske migrerende sammentrekninger. Disse tilsvarer massebevegelsene til tykktarmen hos mennesker og utgjør den viktigste drivkraften for avføring.Hunder er effektene som observeres i mage-tarmkanalen følsomme for blokkering med selektive 5-HT4-reseptorantagonister, og viser dermed at de observerte effektene er utøves gjennom den selektive virkningen på 5-HT4-reseptorer.
Disse farmakodynamiske effektene av prukaloprid ble bekreftet hos mennesker ved manometri som ble brukt hos personer med kronisk forstoppelse i en randomisert, åpen, crossover-studie med blindet vurdering av effekten av prucaloprid 2 mg og et osmotisk avføringsmiddel på kolonmotilitet basert på antall tykktarmskontraksjoner som forplanter seg ved stor amplitude (HAPC, forplantningssammentrekninger med høy amplitude, også kjent som gigantiske migrantkontraksjoner). Sammenlignet med behandlinger for osmotisk forstoppelse, økte den prokinetiske stimuleringen produsert av prukaloprid motiliteten til tykktarmen i den grad uttrykt med antall HAPC i de 12 timene etter administrering av eksperimentelle produktet. Fordelen eller den kliniske relevansen av denne virkningsmekanismen i forhold til andre avføringsmidler er ikke undersøkt.
Klinisk effekt og sikkerhet
Voksen befolkning
Effekten av Resolor ble påvist i tre multisenter, randomiserte, dobbeltblindede, 12-ukers, placebokontrollerte studier hos personer med kronisk forstoppelse (n = 1.279 behandlet med Resolor, 1.124 kvinner, 155 menn). Resolor-dosene ble vurdert hos hver av disse tre studiene var 2 mg og 4 mg en gang daglig. Det primære effektpunktet var prosentandelen (%) av pasientene som oppnådde normalisering av tarmbevegelser definert som et gjennomsnitt på tre eller flere spontane og komplette bevegelser i tarmen (Spontan fullstendig avføring, SCBM) per uke i løpet av 12-ukers behandlingsperiode.
Andelen kvinnelige pasienter for hvilke avføringsmidler ikke ga tilstrekkelig lindring, behandlet med anbefalt 2 mg dose Resolor (n = 458), som oppnådde et gjennomsnitt på ≥ 3 SCBMs per uke var 31. 0% (uke 4) og 24.7 % (uke 12), mot 8,6% (uke 4) og 9,2% (uke 12) i placebogruppen.Klinisk signifikant forbedring av ≥1 SCBM per uke, det viktigste sekundære effektpunktet, ble oppnådd hos 51,0% (uke 4) og 44,2% (uke 12) av pasientene som ble behandlet med 2 mg Resolor, mot 21,7% (uke 4) og 22,6% (uke 12) for placebo.
Effekten av Resolor på spontane avføring (Spontane tarmbevegelser, SBM) var også statistisk bedre enn placebo i andelen pasienter som hadde en økning på ≥1 SBM / uke i løpet av 12-ukers behandlingsperiode. Ved uke 12 hadde 68,3% av pasientene behandlet med Resolor 2 mg en gjennomsnittlig økning på ≥1 SBM / uke, sammenlignet med 37,0% av pasientene som ble behandlet med placebo (s
I alle tre studiene resulterte behandling med Resolor også i betydelige forbedringer i vurderinger av en rekke spesifikke og validerte patologiske symptomer (PAC-SYM, vurdering av pasientforstoppelse), som inkluderer magesymptomer (oppblåsthet, ubehag, smerter og kramper), avføring (ufullstendig avføring, falske alarmer, anstrengelse, overdreven avføringshardhet, utilstrekkelig avføring) og endetarm (smertefulle avføring, svie, blødning / rive), vurdert i uke 4 og uke 12. I uke 4 var prosentandelen pasienter med ≥ 1 forbedring fra baseline på PAC-SYM-subskalaene for abdominale, defekatoriske og rektale symptomer var henholdsvis 41,3%, 41,6% og 31,3% hos pasienter behandlet med Resolor 2 mg, sammenlignet med 26,9%, 24,4% og 22,9% hos placebo- behandlede pasienter. Lignende resultater ble observert i uke 12: henholdsvis 43,4%, 42,9% og 31,7% hos pasienter behandlet med Resolor 2 mg, sammenlignet med 26,9%, 27,2% og 23,4% hos placebobehandlede pasienter (p
I begge evalueringene, i uke 4 og uke 12, ble det også observert en betydelig fordel med hensyn til en rekke parametere angående livskvalitet, for eksempel tilfredshet med behandlingen, tarmvaner og bekymringer, ubehag og fysisk og psykososialt ubehag. I uke 4 var andelen pasienter med ≥1 forbedring fra baseline på subscale Subjective Constipation Assessment-Quality of Life (PAC-QOL) 47,7% hos pasienter behandlet med Resolor 2 mg, sammenlignet med 20, 2% hos pasienter behandlet med placebo. Lignende resultater ble observert i uke 12: 46,9% hos pasienter behandlet med henholdsvis Resolor 2 mg, sammenlignet med 19,0% hos pasienter behandlet med placebo (p
I tillegg ble effekt, sikkerhet og toleranse for Resolor hos mannlige pasienter med kronisk forstoppelse evaluert i en 12 ukers multisenter, randomisert, dobbeltblind, placebokontrollert studie (N = 370). Det primære endepunktet for studien var oppfylt: en statistisk signifikant høyere andel av pasientene i Resolor -gruppen (37,9 %) hadde en gjennomsnittlig ukentlig SCMB ≥ 3, sammenlignet med personer i placebogruppen (17,7 %) (p
Langsiktig studie
Effekten og sikkerheten til Resolor hos pasienter (alder ≥ 18 år) med kronisk forstoppelse ble evaluert i en 24-ukers, multisenter, randomisert, dobbeltblind, placebokontrollert studie (N = 361) av pasienter med en gjennomsnittlig ukentlig frekvens spontan og fullstendig avføring (SCBM) ≥3 (respondere) i løpet av den 24 ukers dobbeltblindede behandlingsfasen var ikke statistisk forskjellig (p = 0,367) mellom behandlingsgruppene med Resolor (25,1%) og placebo (20,7%). Forskjellen mellom behandlingsgruppene fra gjennomsnittlig ukentlig SCBM-frekvens ≥3 var ikke statistisk signifikant for uke 1 til 12, et resultat i kontrast til de andre 5 multisenter, randomiserte, dobbeltblindede, placebokontrollerte studier. 12 ukers varighet som viste effekt av prukaloprid hos voksne pasienter i samme evalueringsperiode. Derfor anses studien å være ufullstendig med hensyn til effekt. Imidlertid støtter helheten av dataene, inkludert de andre dobbeltblindede, placebokontrollerte, 12-ukers studiene effekten av Resolor.Sikkerhetsprofilen til Resolor observert i denne 24-ukers studien er i tråd med det. 12-ukers studier.
Resolor viste ingen rebound -fenomener og var ikke vanedannende.
Fordypning i QT
En grundig QT-studie ble utført for å evaluere effekten av Resolor på QT-intervallet ved terapeutiske (2 mg) og supra-terapeutiske (10 mg) doser, og resultatene ble sammenlignet med effektene av placebo og en positiv kontroll. Denne studien gjorde ikke viste signifikante forskjeller mellom Resolor brukt ved både doser og placebo basert på gjennomsnittlige QT -målinger og en "unormal verdianalyse." Dette bekreftet resultatene av to placebokontrollerte QT-studier. I dobbeltblindede kliniske studier var forekomsten av QT-relaterte bivirkninger og ventrikulære arytmier lave og sammenlignbare med placebogruppen.
Pediatrisk populasjon
Effekten og sikkerheten til Resolor hos pediatriske pasienter (i alderen 6 måneder-18 år) med funksjonell forstoppelse ble evaluert i en 8 ukers, dobbeltblind, placebokontrollert studie (N = 213), etterfulgt av en åpen komparator- kontrollert studie (polyetylenglykol 4000) på 16 uker som varer opptil 24 uker (N = 197). Barn som veide ≤50 kg fikk en startdose på 0,04 mg / kg / dag økte gradvis mellom 0,02 og 0,06 mg / kg / dag ( for maksimalt 2 mg / dag) Resolor oral løsning eller tilsvarende placebo. Barn som veier> 50 kg fikk 2 mg / dag Resolor tabletter eller tilsvarende placebo.
Respons på behandlingen ble definert som et gjennomsnitt på ≥3 spontane tarmbevegelser (SBM) per uke og et gjennomsnittlig antall fekale inkontinensepisoder ≤1 hver 2. uke. Studieresultatene viste ingen forskjeller. Når det gjelder effekt mellom Resolor og placebo: responsrate var henholdsvis 17%og 17,8%(P = 0,9002). Resolor var generelt godt tolerert. Forekomsten av personer med minst 1 bivirkningsdebut under behandling (TEAE) var lik mellom Resolor -gruppen (69,8%) og placebogruppen (60,7%). Samlet sett var sikkerhetsprofilen til Resolor hos barn den samme som hos voksne.
05.2 "Farmakokinetiske egenskaper
Absorpsjon
Prucaloprid absorberes raskt; etter en enkelt oral dose på 2 mg, ble Cmax nådd på 2-3 timer. Absolutt oral biotilgjengelighet er> 90%. Samtidig matinntak påvirker ikke oral biotilgjengelighet av prukaloprid.
Fordeling
Prucalopride er vidt distribuert og har et steady state distribusjonsvolum (Vdss) på 567 liter. Proteinbindingen av prukaloprid er omtrent 30%.
Biotransformasjon
Metabolisme er ikke den viktigste elimineringsveien for prukaloprid. In vitro, menneskelig levermetabolisme er veldig treg, og det finnes bare en minimal mengde metabolitter. I en oral dose med mennesker med radiomerket merket prukaloprid ble det funnet små mengder av syv metabolitter i urin og avføring. Den kvantitativt mest representerte metabolitten i utskillelse, R107504, utgjorde henholdsvis 3,2% og 3,1% av dosen i urin og avføring. Andre metabolitter identifisert og kvantifisert i urin og avføring var R084536 (dannet ved N-dealkylering), tilsvarende 3% av dosen og hydroksyleringsproduktene (3% av dosen) og N-oksidasjon (2% av dosen) uendret virkestoff utgjorde omtrent 92-94% av den totale radioaktiviteten i plasma.R107504, R084536 og R104065 (dannet ved O-demetylering) ble identifisert som mindre plasmametabolitter.
Eliminering
En stor brøkdel av virkestoffet elimineres uendret (60-65% av den administrerte dosen i urinen og ca. 5% i avføringen). Renal utskillelse av uendret prukaloprid innebærer både passiv filtrering og aktiv sekresjon. Plasmaclearance av prukaloprid er i gjennomsnitt 317 ml / minutter. Den terminale halveringstiden er omtrent en dag. Jevn tilstand oppnås innen tre til fire dager. Med en gang daglig behandling med 2 mg. prukaloprid svinger steady-state plasmakonsentrasjoner mellom minimum og maksimumsverdier på henholdsvis 2,5 og 7 ng / ml. Akkumuleringsforholdet etter en enkelt daglig dose varierte fra 1, 9 og 2,3. Farmakokinetikken til prukaloprid er doseproporsjonal både innenfor det terapeutiske området og utover (testet opptil 20 mg). Prucaloprid administrert en gang daglig viser tidsuavhengig kinetikk under langvarig behandling.
Spesielle populasjoner
Befolkningens farmakokinetikk
En populasjonsfarmakokinetisk analyse viste at den tilsynelatende totale clearance av prukaloprid var korrelert med kreatininclearance og at alder, kroppsvekt, kjønn eller rase ikke hadde noen innflytelse.
Pensjonister
Etter en enkelt daglig dose på 1 mg var maksimal plasmakonsentrasjon og AUC for prukaloprid hos eldre personer 26-28% høyere enn hos unge voksne. Denne effekten kan tilskrives redusert nyrefunksjon hos eldre.
Nyresvikt
Sammenlignet med personer med normal nyrefunksjon, var plasmakonsentrasjonen av prukaloprid i gjennomsnitt 25% og 51% høyere hos personer med mild nyreinsuffisiens (ClCR 50-79 ml / minutter) og moderat (ClCR25) -49 ml / minutter). Hos personer med alvorlig nedsatt nyrefunksjon (ClCR ≤ 24 ml / minutter) var plasmakonsentrasjonene 2,3 ganger nivåene som ble funnet hos friske personer (se pkt. 4.2 og 4.4).
Leverinsuffisiens
Ikke-renal eliminering bidrar med omtrent 35% til total eliminering. I en liten farmakokinetisk studie var Cmax og AUC for prukaloprid i gjennomsnitt 10-20% høyere hos pasienter med moderat til alvorlig nedsatt leverfunksjon enn hos friske personer (se pkt. 4.2 og 4.4).
05.3 Prekliniske sikkerhetsdata
Ikke-kliniske data viser ingen spesiell fare for mennesker basert på konvensjonelle studier av sikkerhetsfarmakologi, toksisitet ved gjentatt dosering, gentoksisitet, kreftfremkallende potensial, reproduksjon og utviklingstoksisitet. Et stort antall legemiddelsikkerhetsstudier, utført med spesiell oppmerksomhet på kardiovaskulære parametere, har ikke vist relevante endringer i hemodynamiske og EKG-avledede (QTc) parametere, bortsett fra en beskjeden økning i hjertefrekvens og blodtrykk observert hos bedøvede griser etter intravenøs administrasjon, og en økning i blodtrykk hos bevisste hunder etter intravenøs bolusadministrasjon, som imidlertid ikke ble observert hos bedøvede hunder eller etter oral administrering hos hunder der lignende plasmanivåer oppnås.En neonatal / juvenil subkutan toksisitetsstudie utført på 7-55 dager gamle rotter viste en NOAEL på 100 mg / kg / dag. Eksponeringshastighetene som ble bestemt på grunnlag av AUC0-24t ved NOAEL sammenlignet med dem som ble påvist hos pediatriske personer (behandlet med ca. dose.
06.0 LEGEMIDDELOPPLYSNINGER
06.1 Hjelpestoffer
Kjernen i maispressen
Laktosemonohydrat
Mikrokrystallinsk cellulose
Kolloidalt kiseldioksid
Magnesiumstearat
Tablettbelegg
Hypromellose
Laktosemonohydrat
Triacetin
Titandioksid (E171)
Makrogol
06.2 Uforlikelighet
Ikke relevant.
06.3 Gyldighetsperiode
4 år.
06.4 Spesielle forholdsregler for lagring
Oppbevares i den originale blisterpakningen for å beskytte mot fuktighet.
06.5 Emballasje og innhold i pakningen
Aluminium / aluminium perforerte (kalender) enhetsdoseblister som inneholder 7 tabletter. Hver pakning inneholder 7 x 1, 14 x 1, 28 x 1 eller 84 x 1 filmdrasjerte tabletter.
Det er ikke sikkert at alle pakningsstørrelser blir markedsført.
06.6 Bruksanvisning og håndtering
Ingen spesielle instruksjoner.
07.0 INNEHAVER AV MARKEDSFØRINGSTILLATELSE
Shire Pharmaceuticals Ireland Limited
5 Riverwalk
Citywest Business Campus
Dublin 24
Irland
08.0 NUMMER FOR MARKEDSFØRINGSTILLATELSE
EU/1/09/581/001 (28 tabletter)
EU/1/09/581/003 (7 tabletter)
EU/1/09/581/005 (14 tabletter)
EU/1/09/581/007 (84 tabletter)
041016015
041016027
041016041
041016066
09.0 DATO FOR FØRSTE GODKJENNELSE ELLER FORNYELSE AV GODKJENNINGEN
Dato for første godkjenning: 15. oktober 2009
Dato for siste fornyelse: 6. juni 2014
10.0 DATO FOR REVISJON AV TEKSTEN
05/2015