Årsaken til Duchenne muskeldystrofi er mutasjonen av genet som koder for dystrofin, et protein som er avgjørende for musklenes helse og funksjon.
Duchenne muskeldystrofi begynner å manifestere seg i de første leveårene, og påvirker aktiviteten til lemmenes viktigste muskler; fra dette øyeblikket forverres pasientens tilstand sakte, men gradvis: i barndommen tvinger det dem til rullestol og, mellom ungdomsårene og begynnelsen av voksenlivet, forårsaker det de første hjerte- og luftveisproblemene, hvis utvikling vil være den vanligste dødsårsaken mot 30 år.
Dessverre er det fortsatt ingen kur mot Duchenne muskeldystrofi; de syke kan imidlertid stole på ulike symptomatiske behandlinger som kan lindre symptomene og forlenge levealderen.
Kort påminnelse om genetikk
Hva er DNA?
Inneholdt i cellekjernen er DNA det biologiske makromolekylet som inneholder all informasjon som er nødvendig for riktig utvikling og funksjon av cellene til de levende organismer der den er tilstede.
Hva er kromosomer?
Kromosomer er de strukturelle enhetene der DNA er organisert.
Hver celle i et sunt menneske inneholder 22 par autosomale (eller ikke-kjønn) kromosomer og ett par kjønnskromosomer (disse er et X og et Y, hos menn og to X-er hos kvinner).
Ett kromosom av hvert par av de 23 er fra moren, mens det andre er fra faren.
Hva er gener?
Gener er korte strekninger (eller sekvenser) av DNA med en grunnleggende biologisk betydning: fra dem stammer faktisk molekyler som er grunnleggende for livet: proteiner.
For hvert gen er det to versjoner, kalt alleler, som tilhører den ene til mors kromosom og den andre til farens kromosom.
Hva er en genetisk mutasjon?
Det er en "endring av DNA -sekvensen som danner et gen.
På grunn av denne endringen er det resulterende proteinet enten defekt eller helt fraværende; i begge tilfeller kan effektene være skadelige både for cellens liv, der mutasjonen oppstår, og for organismen som helhet.
Duchenne muskeldystrofi: Hvilke muskler påvirker det?
Duchenne muskeldystrofi påvirker først frivillige muskler i nedre og øvre lemmer som har noe forhold til stammen. Typiske mål for sykdommen er derfor: quadriceps, iliopsoas og baken, med hensyn til underekstremitetene, og deltoidene, pectoral, subscapularis, med hensyn til øvre lemmer.
På et senere tidspunkt strekker DMD seg til pustemuskulaturen og myokardiet, selv om de ikke akkurat er frivillige muskler.
Det skal bemerkes at de første musklene som noensinne har lidd av Duchenne muskeldystrofi, er de i nedre lemmer.
Epidemiologi: Hvor vanlig er Duchenne muskeldystrofi?
For genetiske mekanismer som vil bli diskutert senere, påvirker Duchenne muskeldystrofi hovedsakelig menn og bare sjelden kvinner.
Ifølge epidemiologiske data vil en av hver 3500-6000 menn bli født med DMD.
Det finnes forskjellige former for muskeldystrofi; blant disse er den av Duchenne den vanligste.
Visste du at ...
I Italia ville det være rundt 2000 mennesker med Duchenne muskeldystrofi.
For ytterligere informasjon: Typer muskeldystrofi .Mutasjonen som er ansvarlig for DMD resulterer i fullstendig fravær av dystrofin.
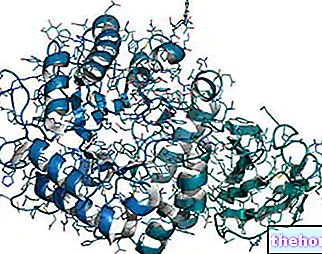
Dystrofin
Dystrofin er et protein som finnes i cellene som utgjør muskelfibrene. Den utfører forskjellige funksjoner, spesielt:
- Den forbinder membranen til muskelfiberen, kalt sarcolemma, til cellemembranen og den ekstracellulære matrisen.
- Det regulerer bevegelsene til kalsiumionen inne i cellen (NB: kalsium er ansvarlig for muskelsammentrekning).
I fravær av dystrofin mislykkes derfor disse prosessene og muskelcellen gjennomgår dødelig oksidativt stress.
Duchenne muskeldystrofi: patogenesen

Fraværet av dystrofin har forskjellige konsekvenser:
- Det skjer en overdreven inntrengning av kalsiumion i sarcolemmaet. Denne hendelsen forårsaker overdreven inntreden av vann i mitokondriene i muskelcellene (myocytter), som "brister".
- Plasmamembranene i muskelceller blir skjøre og brytes lett.
Denne skjørheten i membranene, assosiert med tap av mitokondrier, fører til cellenekrose. - Hendelsene beskrevet ovenfor har en desidert raskere tid enn de cellulære mekanismene som er ansvarlige for å reparere og erstatte kompromitterte myocytter. Dette fører uunngåelig til en gradvis forverring av situasjonen.
- Erstatning av døde myocytter er bindevev (eller fibrotiske) vevs- og fettceller. Denne prosessen fremheves av den eneste tilsynelatende forstørrelsen av noen muskler, en forstørrelse som eksperter definerer med begrepet pseudohypertrofi.
Duchenne muskeldystrofi: Genetikk
Duchenne muskeldystrofi er en sykdom som følger et X-koblet recessivt arvsmønster:
- Koblet til X -kromosomet det betyr at sykdommen avhenger av mutasjonen av et gen som ligger på kjønnskromosom X;
- Resesjon det betyr at begge alleler av det ansvarlige genet må være mutert for at patologien skal være åpenbar.
DMD påvirker hovedsakelig mannlig kjønn av en veldig spesifikk genetisk årsak: mannen har bare ett X -kromosom (det andre er et Y -kromosom), og dermed frata mutasjonen av et gen som er tilstede i det hele organismen det kodede proteinet. Fra det samme gen; kvinnen derimot har to X -kromosomer, og i nærvær av genetiske sykdommer med recessiv arv er mutasjonen av bare en av dem ikke tilstrekkelig til å bestemme patologien (det sunne kromosomet kompenserer for manglene ved mutert).
For at en kvinne skal lide av Duchenne muskeldystrofi, må begge X -kromosomene være mutert i dystrofingenet: dette er en sjelden, men ikke umulig, situasjon som er sett hos omtrent én av 50 000 kvinner.
Faktisk bør det bemerkes at av en rekke komplekse genetiske årsaker, som ikke er beskrevet her, kan DMD også forekomme hos noen kvinner med bare ett mutert X -kromosom.
Duchenne muskeldystrofi: Arv og overføring

Duchenne muskeldystrofi kan være en arvelig sykdom; en genetisk sykdom er en tilstand som skyldes mutasjoner videreført fra en av foreldrene (eller til og med begge deler).
I de fleste tilfeller utløses arvelig DMD av "møtet mellom en frisk mann og en kvinne som er en sunn bærer av sykdommen. En mutasjon i genet som koder for dystrofin.
I en slik situasjon kan bare mannlige barn være syke; døtrene, derimot, kan bare være friske bærere av sykdommen.
Alt dette skjer av årsaken forklart ovenfor, knyttet til det forskjellige antallet X -kromosomer som er tilstede i de to kjønnene.
Her er mer detaljert hva som kan skje når en frisk mann og en frisk kvinne med DMD får et barn:
- DE sønner de har 50% sjanse for å bli syke eller friske. De er syke hvis de arver det muterte X -kromosomet fra moren, mens de er friske hvis de arver X -kromosomet uten mutasjoner fra moren.
- De døtre har en 50% sjanse for å være frisk eller en sunn bærer av sykdommen. De er friske hvis de arver det sunne X -kromosomet fra moren, mens de er friske bærere hvis de mottar det muterte X fra moren.
Som det fremgår, spiller moren alltid en nøkkelrolle i den situasjonen vi undersøker, både med sønner og døtre.
Det skal bemerkes at arvelig Duchenne muskeldystrofi også kan være et resultat av møtet mellom en syk mann og en frisk kvinne som bærer sykdommen.
I denne enestående omstendigheten har mannlige barn fortsatt en 50% sjanse for å være friske eller syke (av samme grunner som i det forrige tilfellet), mens kvinnelige døtre kan være friske bærere av sykdommen eller syke (tar hensyn til det fra deres far døtre arver alltid det muterte X, de er syke hvis de også mottar det muterte X -kromosomet til moren).
Sammenlignet med det første analyserte tilfellet, er denne andre situasjonen desidert sjeldnere, bevis på dette er at én kvinne av 50 000 er født med DMA.
Ervervet Duchenne muskeldystrofi
Selv om det er veldig sjelden, kan Duchenne muskeldystrofi også være en tilstand utviklet like etter unnfangelsen, under embryonal utvikling, som et resultat av en spontan mutasjon.
I dette tilfellet snakker vi om ervervet Duchenne muskeldystrofi.
I ervervede former for DMD er foreldrene både friske, og den mutasjonelle hendelsen er helt utenkelig.
og å løpe; senere sliter han også med å bevege armer og nakke.
Mellom ungdomsårene og begynnelsen av voksenlivet går svakheten videre og påvirker nesten alle musklene, inkludert de ufrivillige som er ansvarlige for pusten og myokardiet.
Det skal bemerkes at noen av pasientene i løpet av sykdommen også utvikler kognitive underskudd og atferdsforstyrrelser.
For ytterligere informasjon: Duchenne muskeldystrofi symptomerDuchenne muskeldystrofi: motoriske symptomer
De karakteristiske motoriske manifestasjonene av Duchenne muskeldystrofi er:
- Forsinkelse av de første trinnene (NB: det kan også forekomme hos friske barn);
- Vanskeligheter med å gå, løpe, hoppe og gå i trapper på grunn av svakhet i musklene i underekstremitetene;
- Turen er svingende, lik den for en "gås" (svingende eller vippende gang);
- Hypostheni i musklene i øvre lemmer og nakke;
- Vanskeligheter med å reise seg fra bakken. Pasienten "klatrer" på seg selv, støtter øvre lemmer og knær (Gowers tegn);
- Utvidelse av kalvene, på grunn av et fenomen som allerede er nevnt kjent som pseudohypertrofi;
- Lumbal skoliose og hyperlordose på grunn av svekkelse av hoftebøyemuskulaturen;
- Kontrakturer på grunn av langvarig immobilitet, etterfulgt av leddforstyrrelser.
Mellom slutten av barndommen og begynnelsen av puberteten er de motoriske vanskelighetene som påvirker underekstremitetene slik at de tvinger pasienten til å bruke rullestol.
Vanligvis, i en alder av 21 år, er DMD -lidelsen lammet fra nakken og ned.
Duchenne muskeldystrofi: kognitive symptomer
Som forventet utvikler noen DMD -pasienter kognitive underskudd og atferdsforstyrrelser over tid.
Disse tilstandene inkluderer for eksempel dysleksi, ADHD (oppmerksomhetsunderskudd hyperaktivitetsforstyrrelse) og korttidshukommelsesunderskudd.
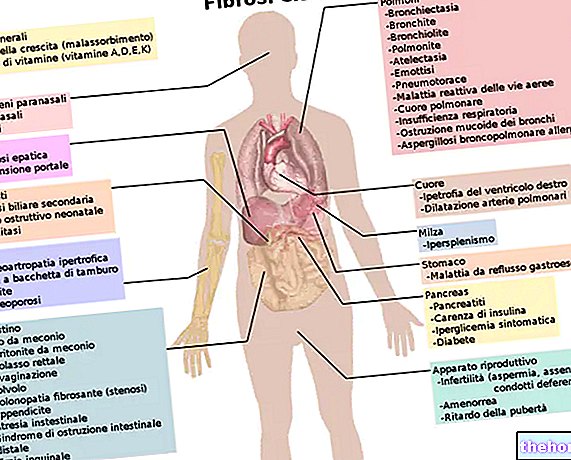
Duchenne muskeldystrofi: komplikasjoner
Vanligvis starter i ungdomsårene eller sent ungdomsår, strekker Duchenne muskeldystrofi seg til hjertets muskler (myokard), noe som resulterer i en form for utvidet kardiomyopati.
Deretter involverer sykdommen også de ufrivillige pustemusklene (membran og intercostale muskler) og musklene som er ansvarlige for tygging; denne ytterligere forverringen, i begynnelsen, disponerer bare for luftveisinfeksjoner (f.eks. lungebetennelse) og forårsaker senere respirasjonssvikt.
Det skal bemerkes at i mellomtiden vises også osteoporose (på grunn av tvungen immobilitet) og gastrointestinale problemer (forstoppelse) knyttet til tap av funksjon av tarmens glatte muskler.
Kardiomyopati og luftveisproblemer utvikler seg til det å være dødelig: vanligvis oppstår faktisk død hos pasienter med Duchenne muskeldystrofi fra hjerte-respiratoriske komplikasjoner.
Dødsalderen for de fleste pasientene er rundt 30 år.
(fysisk undersøkelse);Kreatinkinasemåling
Blodet inneholder et enzym, kreatinkinase (CK eller CPK).
Pasienter med DMD har svært høye doser kreatinkinase, 10 til 100 ganger høyere enn normalt.
Måling av blodkreatinkinase -nivåer er nyttig for å identifisere en unormal tilstand som påvirker musklene, men det er ikke veldig spesifikt: i tillegg til DMD forårsaker faktisk flere andre forhold en betydelig økning i CK.
Muskelbiopsi
Muskelbiopsi er vanligvis det neste trinnet etter kreatinkinasetesten. Analysen av en prøve av muskelvev gjør det faktisk mulig å studere cellene som komponerer det, for å evaluere muskelfibrenes tilstand og for å kvantifisere nivåene av dystrofin.
Fjerning av et stykke muskler innebærer en liten operasjon under lokalbedøvelse.
Muskelbiopsi hos en pasient med DMD: resultatene av analysene
- Fullstendig fravær av dystrofin (lave nivåer kjennetegner andre former for muskeldystrofi).
- Tilstedeværelse av fibro-fettvev, i stedet for muskler (tegn på pseudohypertrofi).
- Muskelfibrene degenererer.
Elektromyografi
Elektromyografi er en diagnostisk prosedyre som tar sikte på å vurdere helsen til muskler og perifere nerver som styrer deres aktivitet.
Basert på bruk av elektroder, nålelektroder og et instrument som kalles en elektromyograf, lar denne testen oss analysere muskelaktivitet som respons på nervestimuli.
Hos mennesker med Duchenne muskeldystrofi viser elektromyografi at musklene ikke reagerer på nerveimpulser som under normale forhold.
Elektromyografi hjelper til med å diagnostisere DMD fordi den skiller patologier på grunn av muskeldysfunksjoner (for eksempel muskeldystrofier) fra sykdommer av nervøs opprinnelse som har konsekvenser for muskelen (f.eks. Motoriske nevronsykdommer).
Genetisk test
Den genetiske analysen av en persons kromosomprofil gjør det mulig å identifisere eventuelle genetiske mutasjoner som påvirker kromosomene.
Den genetiske testen av den kromosomale profilen er vanligvis den diagnostiske undersøkelsen som bekrefter tilstedeværelse eller fravær av Duchenne muskeldystrofi.
Genetisk evaluering kan utføres ikke bare i postnatal fase (etter fødsel), men også før fødsel (prenatal).
For en postnatal test er en blodprøve og påfølgende analyse av blodprøven tilstrekkelig; for en prenatal test, derimot, er det to muligheter: fosterets DNA (som har en liten feilmargin) og analyse av en prøve av fostervann tatt gjennom fostervannsprøve eller CVS (liten risiko for abort).
Fysioterapi, motoraktivitet og ortopediske hjelpemidler
Fysioterapi og regelmessig motorisk aktivitet er avgjørende for å bremse atrofi og svekkelse av musklene.
Å oppmuntre pasienten til å bevege seg, åpenbart så langt som mulig, tjener til å tone muskulaturen og forhindre (eller i det minste utsette) noen komplikasjoner; fysisk trening, faktisk motarbeider osteoporose, forstoppelse og skoliose.
Konstant fysioterapi, postural utdanning og bruk av ortopediske hjelpemidler bevarer, i hvert fall delvis, mobilitet i ledd, sener og muskler.
Husk at ...
Når musklene i underekstremiteten er fullstendig kompromittert, er pasienten med DMD begrenset til en rullestol.
I nærvær av DMD kan bruk av kortikosteroider bidra til å opprettholde pasientens muskelmasse og styrke.
Kortikosteroider som vanligvis brukes hos pasienter med Duchenne muskeldystrofi inkluderer prednisolon og deflazacort.
Behandling og forebygging av komplikasjoner
Når DMD kompliserer helsen til hjertet og respiratoriske muskler, krever pasienten medisiner for å kontrollere utvidet kardiomyopati, en bærbar pusteapparat for mekanisk ventilasjon og noen ganger en pacemaker.
Videre, alltid når sykdommen har nådd dette stadiet, anbefaler leger å skaffe influensavaksine og pneumokokkvaksine for å forhindre de vanligste luftveisinfeksjonene for DMD -pasienter.
I nærvær av alvorlig skoliose er kirurgi nødvendig; det samme gjelder når tyggefunksjonen er fullstendig svekket (gastrostomi).
Vaksiner:
- Anti-influensa
- Anti-pneumokokk
Periodiske kontroller:
- Lungefunksjonstest
- Blodets oksygenivå.
Legemidler til behandling av kardiomyopati:
- Betablokkere;
- Diuretika;
- ACE -hemmere.
Periodiske kontroller:
- Elektrokardiogram;
- Ekkokardiogram;
- Ultralyd.
Medisiner:
- Avføringsmiddel mot forstoppelse.
Kirurgi:
- Gastrostomi, for pasienter med tygging og svelging.
For å gjøre beinene dine "sterkere":
- Administrering av vitamin D og kalsium;
- Eksponering for sollys.
Kirurgi:
- Hoftebøyer tenotomi;
- Spesielt strekking av senene. den achilleo
Godkjente legemidler mot Duchenne muskeldystrofi
- I 2014 godkjente EMA (European Medicines Agency) Ataluren, et stoff som har vist seg å forbedre symptomene på pasienter med cystisk fibrose og Duchenne muskeldystrofi.
Det skal bemerkes at effekten av Ataluren hos DMD -pasienten bare er merkbar når personen fortsatt er i stand til å gå.
Ataluren er ikke akkurat en godkjent medisin for Duchenne muskeldystrofi. - I 2016, i USA, FDA (Food and Drug Administration) godkjente det første spesifikke stoffet mot Duchenne muskeldystrofi: det er Exondys 51, også kjent som eteplirsen.
Denne medisinen ser ut til å kunne fremme produksjonen av dystrofin og dermed motsette seg sykdomsforløpet.
Exondys 51 er kun indisert for bruk hos pasienter med en form for DMD preget av en "endring" av exon 51 i dystrofingenet, som påvirker 13% av de totale tilfellene av Duchenne muskeldystrofi. - I henholdsvis 2019 og 2020 godkjente FDA Vyondys 53 (golodirsen) og Viltepso (viltolarsen), to spesifikke legemidler for behandling av Duchenne muskeldystrofi.
Disse stoffene er kun indikert for bruk hos pasienter med en form for DMD preget av en "endring" av exon 53 i dystrofingenet, som påvirker 8% av de totale tilfellene av Duchenne muskeldystrofi.