Aktive ingredienser: Risedronsyre (Risedronatnatrium)
MEDEOROS 35 mg filmdrasjerte tabletter
Indikasjoner Hvorfor brukes Medeoros? Hva er den til?
MEDEOROS tilhører en gruppe ikke-hormonelle medisiner kalt bisfosfonater som brukes til å behandle bensykdommer (osteoporose). Det virker direkte på beinene, styrker dem og reduserer derfor risikoen for brudd.
Ben er levende vev. Kroppen fjerner kontinuerlig gammelt beinvev og erstatter det med nytt bein.
Postmenopausal osteoporose er en tilstand som utvikler seg hos kvinner etter overgangsalderen når det er svekkelse og tynning av beinene med en påfølgende økt risiko for brudd etter fall eller belastning.
Osteoporose kan også forekomme hos menn av forskjellige årsaker som aldring og / eller et lavt nivå av det mannlige hormonet, testosteron.
De beinene som er mest utsatt for brudd er ryggraden, hoften og håndleddet, selv om alle bein i kroppen kan sprekke. Brudd i forbindelse med osteoporose kan også forårsake ryggsmerter, tap av høyde (vekttap)., Slapphet i ryggen (pukkel) ). Mange pasienter med osteoporose har ingen symptomer og vet ikke engang at de har det.
MEDEOROS brukes til behandling av osteoporose:
- hos postmenopausale kvinner, selv ved alvorlig osteoporose. Reduserer risikoen for brudd på ryggvirvler og hofter
- hos menn med høy risiko for brudd.
Kontraindikasjoner Når Medeoros ikke skal brukes
Ikke ta MEDEOROS:
- hvis du er allergisk mot risedronatnatrium eller noen av de andre innholdsstoffene i dette legemidlet
- hvis du lider av en tilstand som kalles hypokalsemi (lavt kalsiumnivå i blodet);
- hvis du er gravid, mistenker eller planlegger å bli gravid;
- hvis du ammer
- hvis du har alvorlige nyreproblemer.
Forholdsregler for bruk Hva du må vite før du bruker Medeoros
Rådfør deg med lege eller apotek før du bruker MEDEOROS Spesielt:
- Hvis du noen gang har hatt problemer med spiserøret (røret som forbinder munnen din med magen) som forårsaket smerter eller problemer med å svelge mat
- Hvis du ikke klarer å holde overkroppen rett (sittende eller stående) i minst 30 minutter fra du tar tabletten.
- Hvis du har eller nylig har hatt problemer med spiserøret, inkludert Barretts spiserør (en tilstand som er forbundet med endringer i cellene som strekker seg ned i spiserøret);
- Hvis du har forstyrrelser i bein- og mineralmetabolismen (f.eks. Vitamin D -mangel, dysfunksjon i paratyreoideahormon som begge fører til en reduksjon i kalsiumnivået i blodet).
- Hvis du har eller har hatt smerter, hevelse eller nummenhet i kjeven eller en "tung kjeven følelse" eller løsheten av en tann.
- Hvis du blir behandlet av tannlegen din eller planlegger en tannoperasjon, vennligst informer tannlegen din om at du blir behandlet med risedronatnatrium.
- Hvis du lider av en "intoleranse for noen sukkerarter (for eksempel laktose, melkesukker). Pasienter med sjeldne arvelige problemer med galaktoseintoleranse, laktasemangel eller glukose-galaktosemalabsorpsjon bør ikke ta denne medisinen. Legen din vil fortelle deg hva du skal gjøre. gjør mens du tar MEDEOROS hvis du har noen av de ovennevnte tilstandene.
Barn og ungdom
Bruk av natriumrisedronat anbefales ikke til barn og ungdom under 18 år siden data om sikkerhet og effekt er utilstrekkelige.
Interaksjoner Hvilke medisiner eller matvarer kan endre effekten av Medeoros
Medisiner som inneholder en av følgende komponenter reduserer effekten av MEDEOROS når den tas samtidig:
- Fotball
- magnesium
- jern
- aluminium (for eksempel noen blandinger for fordøyelsesbesvær)
Ta disse medisinene minst 30 minutter etter at du har tatt MEDEOROS.
Fortell legen din eller apoteket dersom du bruker, nylig har brukt eller planlegger å bruke andre legemidler.
Med mat og drikke
Det er veldig viktig at du IKKE tar mat eller drikke (unntatt vann fra springen) sammen med MEDEOROS -tabletten slik at den fungerer som den skal. Ikke spesielt ta denne medisinen samtidig som meieriprodukter (for eksempel melk) som de inneholder kalsium (se avsnitt 2 "Andre legemidler og MEDEOROS").
Ta mat og drikke (unntatt vann fra springen) minst 30 minutter etter MEDEOROS -tabletten.
Advarsler Det er viktig å vite at:
Graviditet og amming
IKKE ta MEDEOROS hvis du er gravid, tror at du kan være gravid eller planlegger å bli gravid (se avsnitt 2 "Ikke bruk MEDEOROS"). Den potensielle risikoen forbundet med bruk av risedronatnatrium (aktiv ingrediens i MEDEOROS) hos gravide er ikke kjent.
IKKE ta MEDEOROS hvis du ammer (se avsnitt 2 "Ikke ta MEDEOROS").
MEDEOROS skal bare brukes til behandling av kvinner og menn etter overgangsalderen.
Kjøring og bruk av maskiner
Ingen effekt på evnen til å kjøre bil og bruke maskiner ble observert.
MEDEOROS inneholder laktose
Hvis legen din har fortalt deg at du ikke tåler noen sukkerarter, må du kontakte legen din før du tar denne medisinen (se avsnitt 2 "Advarsler og forsiktighetsregler").
Dose, metode og administrasjonstidspunkt Hvordan du bruker Medeoros: Dosering
Ta alltid dette legemidlet nøyaktig slik legen din har fortalt deg. Rådfør deg med lege eller apotek hvis du er i tvil.
Den anbefalte dosen er 1 MEDEOROS tablett (35 mg risedronatnatrium) en gang i uken. Velg den ukedagen som passer best for aktivitetene dine. Ta en MEDEOROS -tablett en gang i uken på dagen du har valgt.
Boksen har noen rom / mellomrom. Skriv ned ukedagen du har valgt å ta MEDEOROS -nettbrettet. Legg også merke til dagene du skal ta tabletten.
Ta tabletten minst 30 minutter før ditt første måltid på dagen, din første drink, annet enn vann fra springen, eller før andre medisiner.
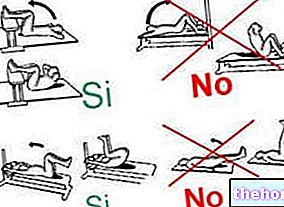
Ta tabletten mens du står oppreist (sittende eller stående) for å unngå halsbrann. Svelg tabletten med minst ett glass vann fra springen (120 ml). Tabletten skal svelges hel. Ikke tygge eller la tabletten smelte i munnen. Ikke legg deg ned i 30 minutter etter at du har svelget tabletten.
Legen din vil fortelle deg om du trenger kalsium og vitamintilskudd hvis de ikke får nok i dietten.
Dersom du har glemt å ta MEDEOROS
Hvis du glemmer å ta tabletten til vanlig tid, ta den den dagen du husker det.
Fortsett å ta en tablett en gang i uken den dagen du opprinnelig valgte.
IKKE ta to tabletter på samme dag som erstatning for en glemt tablett.
Dersom du slutter å ta MEDEOROS
Hvis du slutter å ta MEDEOROS kan du begynne å miste beinmassen. Snakk med legen din før du bestemmer deg for å slutte å ta dette legemidlet.
Spør lege eller apotek hvis du har ytterligere spørsmål om bruken av dette legemidlet.
Overdosering Hva du skal gjøre hvis du har tatt for mye Medeoros
Hvis du eller noen andre ved et uhell har tatt flere MEDEOROS -tabletter enn foreskrevet, drikk et fullt glass melk og kontakt lege umiddelbart.
Bivirkninger Hva er bivirkningene av Medeoros
Som alle andre legemidler kan dette legemidlet forårsake bivirkninger, men ikke alle får det.
Slutt å ta MEDEOROS og kontakt lege umiddelbart hvis du opplever noen av følgende bivirkninger:
- Symptomer på en alvorlig allergisk reaksjon, for eksempel:
- hevelse i ansikt, tunge eller svelg
- problemer med å svelge
- hvetninger (hevet, røde flekker i huden) og pustevansker
- Alvorlige blærende hudreaksjoner, inkludert blemmer.
Fortell legen din umiddelbart hvis du merker følgende bivirkninger: Betennelse i øynene, vanligvis med smerter, rødhet og lysfølsomhet.
Nekrose (ødeleggelse) av beinet i kjeven (osteonekrose) forbundet med forsinket helbredelse og infeksjonsstart, ofte etter tannekstraksjon (se avsnitt 2 "Før du tar MEDEOROS").
Lidelser i spiserøret som svelgesmerter, problemer med å svelge, brystsmerter eller oppstart / forverring av halsbrann.
Imidlertid var de andre bivirkningene som ble observert i kliniske studier vanligvis milde og krevde ikke at pasientene skulle avbryte behandlingen.
Vanlige (rammer 1 til 10 brukere av 100):
- Dyspepsi, kvalme, magesmerter, magekramper eller ubehag i magen, forstoppelse, oppblåsthet, oppblåsthet (økt tarmluft), diaré.
- Smerter i bein, muskler eller ledd.
- Hodepine.
Mindre vanlige (rammer 1 til 10 brukere av 1000):
- Betennelse eller sår i spiserøret (røret som forbinder munnen til magen) som forårsaker problemer med å svelge (se avsnitt 2 "Før du tar MEDEOROS"), betennelse i magen og tolvfingertarmen (den første delen av tarmen som følger mage). - Betennelse i den fargede delen av øyet (iris) (smertefulle røde øyne med mulig nedsatt syn).
Sjeldne (rammer 1 til 10 brukere av 10 000):
- Betennelse i tungen (rød hoven og noen ganger smertefull), innsnevring av spiserøret (røret som forbinder munnen med magen).
- Unormalitet i leverfunksjonstester er rapportert. Disse kan diagnostiseres ved en blodprøve.
Følgende bivirkninger har blitt rapportert etter markedsføring:
Ikke kjent (frekvensen kan ikke estimeres ut fra tilgjengelige data)
- Allergiske hudreaksjoner som elveblest (elveblest), utslett (plutselig rødhet i huden), hevelse i ansikt, lepper, tunge og / eller nakke, svelge- eller pustevansker;
- Alvorlige hudreaksjoner inkludert blemmer under huden; betennelse i små blodkar, preget av håndgripelige røde flekker på huden (leukocytoklastisk vaskulitt);
- En alvorlig tilstand kalt Stevens Johnson syndrom (SJS) med blemmer på hud, munn, øyne og andre fuktige områder av kroppen (kjønnsorganer); en alvorlig sykdom som kalles toksisk epidermal nekrolyse (TEN) som forårsaker rødt utslett på mange deler av kroppen og / eller avskalling av de ytre hudlagene.
- Hårtap
- Allergiske reaksjoner (overfølsomhet).
- Alvorlige leverproblemer, spesielt hvis du blir behandlet med andre medisiner som er kjent for å forårsake leverproblemer.
- Betennelse i øyet forårsaker smerte og rødhet.
Sjelden ved behandlingsstart kan pasientens kalsium- og fosfatnivå i blodet reduseres.
Disse endringene er vanligvis milde og asymptomatiske.
I sjeldne tilfeller kan et uvanlig brudd på lårbenet forekomme spesielt hos pasienter som er i langvarig behandling for osteoporose. Kontakt legen din dersom du opplever smerter, svakhet eller ubehag i lår, hofte eller lyske, da dette kan være en tidlig indikasjon. På en mulig brudd på lårbenet.
Svært sjeldne (forekommer hos opptil 1 av 10 000 personer)
- Snakk med legen din dersom du har ørepine, øreutladning og / eller ørebetennelse. Disse episodene kan være tegn på beinskader i øret.
Rapportering av bivirkninger
Rådfør deg med lege eller apotek dersom du får bivirkninger, inkludert mulige bivirkninger som ikke er nevnt i dette pakningsvedlegget. Du kan også melde fra om bivirkninger direkte til Det italienske legemiddelverkets nettsted: www.agenziafarmaco.gov.it/it/responsabili. Ved å rapportere bivirkninger kan du bidra med mer informasjon om sikkerheten til dette legemidlet.
Utløp og oppbevaring
Hold denne medisinen utilgjengelig for barn.
Denne medisinen krever ingen spesielle oppbevaringsbetingelser.
Ikke bruk dette legemidlet etter utløpsdatoen som er angitt på esken. Utløpsdatoen refererer til den siste dagen i den måneden.
Ikke kast medisiner i avløpsvann eller husholdningsavfall. Spør apoteket om hvordan du skal kaste medisiner du ikke bruker lenger. Dette vil bidra til å beskytte miljøet.
Annen informasjon
Hva MEDEOROS inneholder
- Den aktive ingrediensen er risedronatnatrium. Hver tablett inneholder 35 mg risedronatnatrium (som risedronatnatriumhemipentahydrat).
- Andre innholdsstoffer er: Kjerne: Mikrokrystallinsk cellulose, crospovidon, magnesiumstearat, laktosemonohydrat.
- Belegg: rødt jernoksid, gult jernoksid, vannfritt kolloidalt silika, titandioksid, makrogol 400, makrogol 8000, hypromellose, hydroksypropylcellulose.
Beskrivelse av hvordan MEDEOROS ser ut og innholdet i pakningen
MEDEOROS er runde, lyse oransje filmdrasjerte tabletter med en diameter på 9 mm.
De er tilgjengelige i blisterpakninger som inneholder 4 tabletter.
Kildepakningsvedlegg: AIFA (Italian Medicines Agency). Innhold publisert i januar 2016. Informasjonen som er tilstede er kanskje ikke oppdatert.
For å få tilgang til den mest oppdaterte versjonen, er det lurt å gå til nettstedet til AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttig informasjon.
01.0 LEGEMIDLETS NAVN
MEDEOROS 35 MG -TABLETTER LAGET MED FILM
02.0 KVALITATIV OG KVANTITATIV SAMMENSETNING
Hver tablett inneholder:
Aktiv ingrediens: 35 mg risedronatnatrium (som 40,2 mg risedronatnatriumhemipentahydrat)
Hjelpestoff med kjente effekter: laktose.
For fullstendig liste over hjelpestoffer, se pkt.6.1.
03.0 LEGEMIDDELFORM
Filmdrasjerte tabletter.
Runde, lyse oransje tabletter med en diameter på 9 mm.
04.0 KLINISK INFORMASJON
04.1 Terapeutiske indikasjoner
Behandling av postmenopausal osteoporose for å redusere risikoen for vertebrale brudd Behandling av manifest postmenopausal osteoporose for å redusere risikoen for hoftebrudd (se pkt. 5.1).
Behandling av osteoporose hos menn med høy risiko for brudd (se pkt.5.1).
04.2 Dosering og administrasjonsmåte
Dosering
Den anbefalte dosen for voksne er en 35 mg tablett tatt oralt en gang i uken. Tabletten skal tas samme dag hver uke.
Pediatrisk populasjon:
Bruk av risedronatnatrium anbefales ikke til barn under 18 år på grunn av utilstrekkelige data om sikkerhet og effekt (se også pkt.5.1).
Eldre pasienter:
Ingen dosejustering er nødvendig da biotilgjengelighet, distribusjon og eliminering hos eldre personer (> 60 år) ble funnet å være lik de hos yngre personer. Dette ble også påvist hos svært eldre pasienter, dvs. 75 år. Og senere i postmenopausen befolkning.
Nedsatt nyrefunksjon:
Ingen dosejustering er nødvendig hos pasienter med mild til moderat nedsatt nyrefunksjon. Bruk av natriumrisedronat er kontraindisert hos pasienter med alvorlig nedsatt nyrefunksjon (kreatininclearance mindre enn 30 ml / min) (se pkt. 4.3 og 5.2).
Administrasjonsmåte
Absorpsjonen av risedronatnatrium påvirkes av mat, og derfor bør pasienter ta risedronatnatrium for å sikre tilstrekkelig absorpsjon:
• før frokost: minst 30 minutter før inntak av den første maten, andre medisiner eller drikkevarer på dagen (unntatt vann fra springen).
Pasienter bør informeres om at hvis de glemmer å ta en MEDEOROS 35 mg tablett, bør de ta den den dagen de husker det. Pasienter bør deretter fortsette å ta en tablett per uke den dagen tabletten vanligvis tas. To tabletter bør ikke tas samme dag.
Tabletten skal svelges hel og ikke oppløses i munnen eller tygges. For å lette passasjen av spiserøret, ta risedronatnatrium med et glass vann fra springen (≥120 ml) mens torso holdes oppreist (stående eller sittende). Når tabletten er inntatt, bør pasientene unngå sengetid i 30 minutter (se pkt. 4.4).
Kalsium og vitamin D -tilskudd bør vurderes ved utilstrekkelig diettinntak.
Den optimale varigheten av bisfosfonatbehandling for osteoporose er ikke fastslått.Behovet for fortsatt behandling bør vurderes regelmessig hos hver enkelt pasient ut fra de potensielle fordelene og risikoene, spesielt etter 5 eller flere års bruk.
04.3 Kontraindikasjoner
Overfølsomhet overfor virkestoffet eller overfor noen av hjelpestoffene listet opp i pkt.6.1.
Hypokalsemi (se pkt. 4.4).
Graviditet og amming.
Alvorlig nedsatt nyrefunksjon (kreatininclearance
04.4 Spesielle advarsler og passende forholdsregler for bruk
Mat, drikke (unntatt vann fra springen) og legemidler som inneholder flerverdige kationer (som kalsium, magnesium, jern og aluminium) forstyrrer absorpsjonen av bisfosfonater og bør ikke tas samtidig med natriumrisedronat (se pkt. 4.5). For å oppnå ønsket effekt må administrasjonsinstruksjonene følges nøye (se pkt.4.2).
Effekten av bisfosfonater ved behandling av postmenopausal osteoporose er relatert til tilstedeværelsen av redusert bentetthet og / eller forekomst av brudd.
Eldre alder eller kliniske risikofaktorer for brudd alene berettiger ikke å starte behandling med osteoporose med et bisfosfonat.
Det er begrenset bevis som støtter effekten av bisfosfonater inkludert risedronatnatrium hos svært eldre kvinner (over 80 år) (se pkt.5.1).
Bisfosfonater har vært assosiert med esophagitt, gastritt, esophageal sår og gastroduodenalsår. Derfor bør det utvises forsiktighet:
• hos pasienter som tidligere har hatt esophageal lidelser som forårsaker forsinket esophageal transitt eller gastrisk tømming, for eksempel innsnevring eller achalasi;
• hos pasienter som ikke klarer å holde torso oppreist i minst 30 minutter fra de tok tabletten;
• hvis risedronatnatrium brukes til pasienter med nåværende eller nylige problemer med øvre mage -tarmkanal eller spiserøret (inkludert Barretts spiserør).
Leger bør understreke overfor pasientene viktigheten av å følge administrasjonsinstruksjonene og være oppmerksomme på eventuelle tegn eller symptomer som indikerer en mulig spiserørreaksjon.Pasienter bør informeres om at hvis de utvikler symptomer på esophageal irritasjon som dysfagi, svelging, retrosternal smerte eller begynnelse / forverring av halsbrann, bør søke øyeblikkelig legehjelp.
Hypokalsemi bør korrigeres før behandling med natriumrisedronat startes. Det er også nødvendig å korrigere andre forstyrrelser i bein- og mineralmetabolismen (f.eks. Parathyroidea dysfunksjon, hypovitaminose D) når behandling med risedronatnatrium startes.
Osteonekrose i kjeven, vanligvis forbundet med tanntrekking og / eller lokal infeksjon (inkludert osteomyelitt), er rapportert hos kreftpasienter behandlet med behandlinger inkludert bisfosfonater administrert primært intravenøst. Mange av disse pasientene ble de også behandlet med cellegift og kortikosteroider. Osteonekrose av kjeven er også rapportert hos pasienter med osteoporose som blir behandlet med orale bisfosfonater.
Før du starter behandling med bisfosfonater hos pasienter med samtidige risikofaktorer (som kreft, cellegift, strålebehandling, kortikosteroider, dårlig munnhygiene) bør behovet for en tannundersøkelse med passende forebyggende tannbehandling vurderes.
Under behandlingen bør disse pasientene om mulig unngå invasive tannbehandlinger. Hos pasienter som har utviklet osteonekrose i kjeven og / eller kjeven under bisfosfonatbehandling, kan tannkirurgi forverre tilstanden. For pasienter som trenger tannkirurgi, er det ingen tilgjengelige data som tyder på at seponering av bisfosfonat reduserer risikoen for osteonekrose i kjeven.
Den kliniske vurderingen av legen må være veiledende i behandlingsprogrammet for hver pasient, basert på den individuelle vurderingen av risiko / nytte -forholdet.
Osteonekrose i den eksterne hørselskanalen er rapportert i forbindelse med bruk av bisfosfonater, hovedsakelig i forbindelse med langvarige behandlinger. som infeksjon eller traume. Osteonekrose i den ytre hørselskanalen bør vurderes hos pasienter behandlet med bisfosfonater som har øresymptomer, inkludert kroniske øreinfeksjoner.
Atypiske brudd på lårbenet
Atypiske subtrokanteriske og akselfrakturer i lårbenet er rapportert, hovedsakelig hos pasienter på langvarig bisfosfonatbehandling mot osteoporose Disse korte tverrgående eller skrå bruddene kan forekomme hvor som helst i lårbenet fra like under den mindre trochanteren til over den suprakondylære linjen. Disse bruddene oppstår spontant eller etter minimalt traume, og noen pasienter opplever smerter i lår eller lyske, ofte assosiert med avbildningsfunn og radiografiske tegn på stressfrakturer, uker eller måneder før utbruddet av stressbrudd.en fullstendig lårbensbrudd. Brudd er ofte bilaterale; Derfor bør bisfosfonatbehandlede pasienter som har pådratt seg et lårbenaksfraktur, undersøkes det kontralaterale lårbenet. Begrenset helbredelse av disse bruddene er også rapportert. Hos pasienter med mistanke om atypisk lårbenbrudd, bør det vurderes å avbryte behandlingen med bisfosfonater i påvente av en vurdering av pasienten basert på individuell nytterisiko.
Under behandling med bisfosfonater bør pasientene rådes til å rapportere smerter i lår, hofte eller lyske, og enhver pasient som viser slike symptomer, bør vurderes for en ufullstendig brudd på lårbenet.
Dette stoffet inneholder laktose. Pasienter med sjeldne arvelige problemer med galaktoseintoleranse, laktasemangel eller glukose-galaktosemalabsorpsjon bør ikke ta dette legemidlet.
04.5 Interaksjoner med andre legemidler og andre former for interaksjon
Ingen interaksjonsstudier med andre behandlinger har blitt utført, men klinisk relevante interaksjoner med andre legemidler har ikke blitt observert i kliniske studier.I fase III -studier av risedronatnatrium ved behandling av osteoporose tok henholdsvis 33% og 45% av pasientene acetylsalisylsyre syre eller andre ikke-steroide antiinflammatoriske legemidler (NSAIDs). I fase III-studien med ukentlig dosering, fikk 57% og 40% av postmenopausale pasienter henholdsvis acetylsalisylsyre eller andre ikke-steroide antiinflammatoriske legemidler. Blant pasienter som regelmessig ble behandlet med acetylsalisylsyre eller NSAID (3 eller flere dager i uken), var forekomsten av øvre gastrointestinale bivirkninger hos pasienter behandlet med risedronatnatrium lik den i kontrollgruppen.
Hvis det anses hensiktsmessig, kan risedronatnatrium brukes samtidig med østrogenerstatningsterapi (kun for kvinner).
Samtidig bruk av legemidler som inneholder flerverdig kation (f.eks. Kalsium, magnesium, jern og aluminium) forstyrrer absorpsjonen av risedronatnatrium (se pkt. 4.4).
Risedronatnatrium metaboliseres ikke systemisk, induserer ikke cytokrom P-450 enzymer og har lav proteinbinding.
04.6 Graviditet og amming
Svangerskap
Det er utilstrekkelige data om bruk av risedronatnatrium hos gravide. Studier på dyr har vist reproduksjonstoksisitet (se pkt. 5.3) Den potensielle risikoen for kvinner er ukjent.
Foringstid
Dyrestudier indikerer at en liten mengde natriumrisedronat passerer over i morsmelk.
Risedronatnatrium bør ikke gis til gravide eller ammende kvinner.
04.7 Påvirkning av evnen til å kjøre bil og bruke maskiner
Ingen effekt på evnen til å kjøre bil og bruke maskiner ble observert.
04.8 Bivirkninger
Risedronatnatrium har blitt studert i kliniske fase III -studier med mer enn 15 000 pasienter.
De fleste bivirkningene som ble sett i kliniske studier var milde eller moderate i alvorlighetsgrad og krevde vanligvis ikke seponering av behandlingen.
Uønskede effekter som oppstod under kliniske fase III -studier med kvinner med postmenopausal osteoporose behandlet i opptil 36 måneder med risedronatnatrium i en dose på 5 mg / dag (n = 5020) eller med placebo (n = 5048), og vurderes muligens eller sannsynligvis relatert til risedronatnatrium, er oppført ved hjelp av følgende definisjon (forekomsten versus placebo er angitt i parentes):
Svært vanlige (≥1 / 10); vanlig (≥1 / 100;
Nervesystemet lidelser:
Vanlig: hodepine (1,8% mot 1,4%)
Øyesykdommer:
Mindre vanlige: iritt *
Gastrointestinale lidelser:
Vanlige: forstoppelse (5,0% mot 4,8%), dyspepsi (4,5% mot 4,1%), kvalme (4,3% mot 4,0%), magesmerter (3,5% mot 3,3%), diaré (3,0% vs. 2,7) %)
Mindre vanlige: gastritt (0,9% vs. 0,7%), øsofagitt (0,9% vs. 0,9%), dysfagi (0,4% vs. 0,2%), duodenitt (0,2% vs. 0,1%), øsofagus sår (0,2% vs. 0,2) %)
Sjelden: glossitt (esophageal stricture (
Muskel -skjelett- og bindevevssykdommer:
Vanlig: muskuloskeletale smerter (2,1% mot 1,9%).
Diagnostiske tester:
Sjeldne: leverfunksjonstestavvik *
* Ingen relevant forekomst fra kliniske fase III -studier med osteoporose; frekvensen er basert på uønskede / laboratorie- / oppfordringsdata fra tidligere kliniske studier.
I en 1-årig, dobbeltblind, multisenterstudie som sammenlignet risedronat 5 mg daglig (n = 480) og risedronatnatrium 35 mg en gang i uken (n = 485) hos postmenopausale kvinner med osteoporose, var den generelle toleransen og sikkerhetsprofilene like. Følgende ytterligere bivirkninger som etterforskeren anser som muligens eller sannsynligvis narkotikarelatert (høyere forekomst i gruppen med risedronat 35 mg enn i gruppen med 5 mg risedronatnatrium) ble rapportert: gastrointestinale forstyrrelser (1,6% mot 1,0%) og smerter ( 1,2% mot 0,8%).
I en 2-årig multisenterstudie utført hos menn med osteoporose, var de generelle sikkerhets- og tolerabilitetsprofilene mellom den aktive terapigruppen og placebogruppen like. Bivirkningene samsvarer med de som tidligere er sett hos kvinner.
LaboratorieparametereInnledende milde, forbigående og asymptomatiske reduksjoner i serumkalsium og fosfat har blitt observert hos noen pasienter.
Følgende ytterligere bivirkninger er rapportert fra markedsføring: (frekvens ikke kjent):
Øyesykdommer:
iritt, uveitt.
Muskel -skjelett- og bindevevssykdommer:
osteonekrose i underkjeven og / eller overkjølen.
Hud og subkutant vev:
hud- og overfølsomhetsreaksjoner, inkludert angioødem, generalisert utslett, urticaria og bullous hudreaksjoner og leukocytoklastisk vaskulitt, inkludert noen alvorlige isolerte tilfeller av Stevens Johnson syndrom og toksisk epidermal nekrolyse.
Hårtap
Forstyrrelser i immunsystemet:
anafylaktiske reaksjoner.
Hepatobiliære lidelser:
alvorlig leversykdom. I de fleste tilfellene som ble rapportert, ble pasienter også behandlet med andre produkter som er kjent for å forårsake leversykdom.
Følgende reaksjoner har blitt rapportert etter markedsføring (sjelden frekvens): Atypiske subtrokanteriske og diafysale brudd på lårbenet (bisfosfonatklasse bivirkning).
Svært sjelden: osteonekrose i den ytre hørselskanalen (bivirkning for bisfosfonatklassen).
Rapportering av mistenkte bivirkninger
Rapportering av mistenkte bivirkninger som oppstår etter godkjenning av legemidlet er viktig, ettersom det muliggjør kontinuerlig overvåking av nytte / risiko -forholdet til legemidlet Helsepersonell bes rapportere eventuelle mistenkte bivirkninger via Det italienske legemiddelkontoret. på adressen http://www.agenziafarmaco.gov.it/it/responsabili.
04.9 Overdosering
Ingen spesifikke data er tilgjengelige om behandling av tilfeller av overdose med risedronatnatrium.
Ved overdosering kan det forventes redusert serumkalsium. Noen av disse pasientene kan også ha tegn og symptomer på hypokalsemi.
Melk eller antacida som inneholder magnesium, kalsium eller aluminium bør gis for å binde risedronat og redusere absorpsjonen.I tilfeller av overdosering kan mageskylling vurderes for å fjerne ikke -absorbert risedronatnatrium.
05.0 FARMAKOLOGISKE EGENSKAPER
05.1 Farmakodynamiske egenskaper
Farmakoterapeutisk kategori: Bisfosfonater, ATC-kode M05BA07.
Risedronatnatrium er et pyridinylbisfosfonat som fester seg til hydroksyapatitt i beinet og hemmer beinresorpsjon av osteoklaster. Benomsetningen reduseres mens osteoblastisk aktivitet og benmineralisering opprettholdes.I prekliniske studier har natriumrisedronat vist en kraftig anti-osteoklastisk og antiresorpsjon som resulterer i en doseavhengig økning i benmasse og biomekanisk styrke av bein. Aktiviteten til risedronatnatrium ble bekreftet ved målinger av biokjemiske indekser for beinomsetning under farmakodynamiske og kliniske studier.I studier av postmenopausale kvinner ble reduksjoner i biokjemiske indekser for beinomsetning observert i løpet av den første måneden og nådde maksimumsnivået innen 3-6 måneder Nedgangen i disse indeksene var lik med Risedronate 35 mg per uke og Risedronate 5 mg / dag etter 12 måneder.
I en studie hos menn med osteoporose ble reduksjoner i biokjemiske indekser for beinomsetning observert så tidlig som 3 måneder og fortsatte å bli observert etter 24 måneder.
Terapi og forebygging av postmenopausal osteoporose :
Mange risikofaktorer, inkludert lav benmasse, lav beinmineraltetthet, tidlig overgangsalder, røyking og familiehistorie av osteoporose er forbundet med postmenopausal osteoporose.Den kliniske konsekvensen av osteoporose er økt forekomst av brudd. Risikoen for brudd øker med økende risikofaktorer.
Basert på effektene på BMD i lumbal ryggrad, ble Risedronate 35 mg / uke (n = 485) vist å være ekvivalent med Risedronate 5 mg / dag (n = 480) i en multisenter, dobbeltblind studie av ett års varighet , hos postmenopausale kvinner med osteoporose.
Den daglige doseringen av det kliniske utviklingsprogrammet for risedronatnatrium evaluerte effekten av risedronatnatrium på risikoen for hofte- og vertebrale frakturer og inkluderte både tidlige og sene postmenopausale kvinner med eller uten brudd. Doser på 2 ble evaluert., 5 og 5 mg per dag og alle grupper, inkludert kontroller, mottok kalsium og vitamin D (hvis grunnlinjenivåene var lave). Den absolutte og relative risikoen for nye vertebrale og hoftebrudd ble beregnet ved "bruk av en" analyse "tid til første arrangement'.
• To placebokontrollerte studier (n = 3.661) inkluderte postmenopausale kvinner under 85 år med vertebrale frakturer ved baseline.Risedronatnatrium 5 mg daglig gitt i 3 år resulterte i en reduksjon i risikoen for nye vertebrale brudd sammenlignet med kontrollen Hos kvinner med minst 2 vertebrale brudd var den relative risikoreduksjonen av nye brudd 49% (forekomsten av nye vertebrale frakturer med risedronatnatrium var 18,1% og med placebo 29%), hos de med minst 1 brudd var denne reduksjonen 41%, (forekomsten av nye vertebrale brudd med risedronat var 11,3%, mens det med placebo var 16,3%). Behandlingseffekten ble observert allerede ved slutten av det første behandlingsåret.Fordelene ble også påvist hos kvinner med flere brudd ved baseline.Risedronatnatrium 5 mg daglig redusert årlig tap sammenlignet med kontrollgruppen.
• Ytterligere to placebokontrollerte studier inkluderte postmenopausale kvinner over 70 år med eller uten virvelfrakturer ved grunnlinjen. Kvinner i alderen 70-79 år med skjelettlårhals BMD T-score ble registrert for hoftebrudd eller basert på redusert mineraltetthet av lårhals. Statistisk sett ble effekten av risedronatnatrium versus placebo bare oppnådd når de to gruppene som ble behandlet med 2,5 og 5 mg ble kombinert. Følgende resultater er basert på post-post analyse av pasientundergrupper bare valgt fra kliniske tilfeller eller på nåværende definisjon av osteoporose:
• I en undergruppe av pasienter med lårhals BMD T-score ≤-2,5 SD (NHANES III) og med minst én vertebral fraktur ved baseline reduserte risedronatnatrium administrert i tre år risikoen for hoftebrudd. Hoftebrudd i 46% av tilfellene sammenlignet med kontrollgruppen (forekomsten av hoftebrudd i 2,5 og 5 mg risedronat -natriumgruppene var 3,8%, 7,4% med placebo).
• Dataene tyder på at mer begrenset beskyttelse er tydelig hos eldre pasienter (≥80 år). Dette kan være en konsekvens av den økte betydningen av ikke-skjelettfaktorer for hoftebrudd gjennom årene. I disse studiene belyste den sekundære endepunktsanalysen den reduserte risikoen for nye vertebrale brudd hos pasienter med redusert lårhals BMD uten vertebrale frakturer og hos pasienter med redusert lårhals BMD med eller uten vertebrale brudd.
• Risedronatnatrium 5 mg daglig gitt i 3 år økte beinmineraltettheten (BMD) i korsryggen, lårhalsen, trochanter og håndleddet sammenlignet med kontrollgruppen og forhindret bentap i den distale tredjedelen av radioen.
• En rask reduksjon i undertrykkende effekter av risedronatnatrium på beinomsetningshastigheten ble observert i "året etter avsluttet behandling etter tre års behandling med risedronatnatrium 5 mg daglig."
• Benbiopsier utført på postmenopausale kvinner behandlet med risedronatnatrium 5 mg daglig i 2-3 år bekreftet den forventede moderate nedgangen i beinomsetningen. Benvev under risedronat -natriumbehandling ble funnet å ha en normal lamellstruktur og beinmineraliseringshastighet. Disse dataene, sammen med den reduserte forekomsten av osteoporotiske vertebrale brudd hos kvinner med osteoporose, ser ut til å bekrefte fraværet av skadelige effekter på beinkvaliteten.
Endoskopiske målinger utført på en rekke pasienter, både på risedronatnatrium og i kontrollgruppen, som lider av ulike moderate til alvorlige gastrointestinale lidelser, avslørte ikke esophageal, magesår eller duodenalsår relatert til behandlingen, selv om tilfeller av duodenitt ble observert uvanlig i risedronat -natriumgruppen.
Osteoporose terapi hos menn
Risedronatnatrium 35 mg en gang i uken ble vist å være effektivt hos menn med osteoporose (i alderen 36 til 84 år) i en 2-årig, dobbeltblind, placebokontrollert studie hos 284 pasienter (risedronatnatrium 35 mg n = 191). Alle pasientene fikk tilskudd av kalsium og vitamin D.
Økninger i BMD ble sett så tidlig som 6 måneder etter initiering av behandling med risedronatnatrium. Risedronatnatrium 35 mg én gang i uken ga gjennomsnittlig økning i BMD i korsryggen, lårhalsen, trochanter og hoften sammenlignet med placebo etter 2 års behandling. Effekten mot brudd ble ikke påvist i denne studien.
Effekten på bein (økt BMD og reduserte biokjemiske markører for beinomsetning) av risedronatnatrium er lik hos menn og kvinner.
Pediatrisk populasjon
Sikkerhet og effekt av risedronatnatrium evalueres i en pågående studie hos pediatriske pasienter fra 4 år til under 16 år med osteogenesis imperfecta. Etter avsluttet randomisert, dobbeltblind, placebokontrollert fase, som varer i ett år, en statistisk signifikant økning i BMD i lumbale ryggraden ble påvist i risedronatgruppen kontra placebogruppen; Imidlertid ble en økning i antallet på minst 1 ny morfometrisk vertebral brudd (radiografisk vurdert) funnet i risedronatgruppen sammenlignet med placebo. , resultatene støtter ikke bruk av risedronatnatrium hos pediatriske pasienter med osteogenesis imperfecta.
05.2 Farmakokinetiske egenskaper
Absorpsjon
Absorpsjon av en oral dose er relativt rask (Tmax ~ 1 time) og er doseuavhengig over doseområdet som er undersøkt (enkeltdosestudie 2,5 til 30 mg; flerdosestudier 2,5 til 5 mg / dag og opptil 50 mg / uke). Den orale biotilgjengeligheten til tabletten er i gjennomsnitt 0,63% og reduseres når risedronatnatrium administreres sammen med mat. Biotilgjengeligheten var lik hos menn og kvinner.
Fordeling: gjennomsnittlig distribusjonsvolum ved steady state hos mennesker er 6,3 l / kg.
Brøkdelen av legemidlet bundet til plasmaproteiner er omtrent 24%.
Biotransformasjon:
Det er ingen bevis for at natriumrisedronat metaboliseres systemisk.
Eliminering
Omtrent halvparten av den absorberte dosen elimineres i urinen innen 24 timer, mens 85% av en intravenøs dose elimineres i urinen etter 28 dager. Gjennomsnittlig renal clearance er 105 ml / min og total clearance er 122 ml. / Min: forskjellen skyldes sannsynligvis clearance på grunn av absorpsjon til bein Renal clearance er ikke konsentrasjonsavhengig og det er en lineær sammenheng mellom renal clearance og kreatininclearance Uabsorbert risedronatnatrium elimineres uendret via avføringen Etter oral administrering viser konsentrasjon-tidskurven tre eliminasjonsfaser med en terminal halveringstid på 480 timer.
Spesielle populasjoner
Eldre pasienter:
Ingen dosejustering er nødvendig.
Pasienter behandlet med acetylsalisylsyre / NSAID:
Blant pasienter som ble behandlet regelmessig (tre eller flere dager i uken) med acetylsalisylsyre eller NSAID, var forekomsten av bivirkninger i det øvre mage -tarmkanalen i risedronatnatrium lik den i kontrollgruppen.
05.3 Prekliniske sikkerhetsdata
Doseavhengige hepatotoksiske effekter av risedronatnatrium, hovedsakelig som en økning i enzymer, med histologiske endringer hos rotter, ble observert i toksikologiske studier på rotter og hunder. Den kliniske relevansen av disse observasjonene er ukjent. Testikeltoksisitet dukket opp hos rotter og hunder ved eksponeringer som ble ansett som overstiger den terapeutiske eksponeringen hos mennesker. Hos gnagere har doseavhengig irritasjon av de øvre luftveiene ofte blitt notert. Lignende effekter har blitt rapportert med andre bisfosfonater. Effekter på nedre luftveier har blitt observert i langtidsstudier på gnagere, men den kliniske relevansen av disse funnene er uklar. I reproduksjonstoksisitetsstudier for nær kliniske eksponeringer ble det observert endringer i ossifikasjon på sternal- og / eller kranialnivå hos fostre av behandlede rotter og hypokalsemi og dødelighet hos behandlede hunner som fødte. Det er ingen tegn på teratogenese på fødselstidspunktet . dose på 3,2 mg / kg / dag hos rotter og 10 mg / kg / dag hos kaniner, selv om data bare er tilgjengelig om et begrenset antall kaniner. Maternell toksisitet forhindret studier av høyere doser. om gentoksisitet og karsinogenese har ikke vist noen særlig risiko for mennesker.
06.0 LEGEMIDDELOPPLYSNINGER
06.1 Hjelpestoffer
Kjerne: Mikrokrystallinsk cellulose, crospovidon, magnesiumstearat, laktosemonohydrat,
Belegg: rødt jernoksid, gult jernoksid, vannfritt kolloidalt silika, titandioksid, makrogol 400, makrogol 8000, hypromellose, hydroksypropylcellulose.
06.2 Uforlikelighet
Ikke relevant.
06.3 Gyldighetsperiode
3 år.
06.4 Spesielle forholdsregler for lagring
Denne medisinen krever ingen spesielle oppbevaringsbetingelser.
06.5 Emballasje og innhold i pakningen
Ugjennomsiktig blister av PVC / PVDC / aluminium i en pappeske
Pakning: 4 tabletter
06.6 Bruksanvisning og håndtering
Ingen spesielle instruksjoner.
07.0 INNEHAVER AV MARKEDSFØRINGSTILLATELSE
FENIX PHARMA SOC. KOOPERATIV
Via Ercolano Salvi n. 18
00143 Roma
Italia
08.0 NUMMER FOR MARKEDSFØRINGSTILLATELSE
AIC n. 040044012 "MEDEOROS 35 mg filmdrasjerte tabletter - 4 tabletter"
09.0 DATO FOR FØRSTE GODKJENNELSE ELLER FORNYELSE AV GODKJENNINGEN
Dato for første godkjenning: 20. juni 2011
Siste fornyelsesdato:
10.0 DATO FOR REVISJON AV TEKSTEN
Februar 2016