Aktive ingredienser: Golimumab
Simponi 50 mg injeksjonsvæske, oppløsning i ferdigfylt penn
Simponi pakningsinnsatser er tilgjengelige for pakningsstørrelser:- Simponi 50 mg injeksjonsvæske, oppløsning i ferdigfylt penn
- Simponi 100 mg injeksjonsvæske, oppløsning i ferdigfylt penn
Indikasjoner Hvorfor brukes Simponi? Hva er den til?
Simponi inneholder et virkestoff som kalles golimumab.
Simponi tilhører en gruppe legemidler som kalles 'TNF -blokkere'. Det brukes hos voksne for behandling av følgende inflammatoriske sykdommer:
- Leddgikt
- Psoriasisartritt
- Aksial spondylartritt, inkludert ankyloserende spondylitt og ikke-radiografisk aksial spondylartritt
- Ulcerøs kolitt
Hos barn som veier minst 40 kg, brukes Simponi til behandling av polyartikulær juvenil idiopatisk artritt.
Simponi virker ved å blokkere virkningen av et protein som kalles "tumornekrosefaktor alfa" (TNF-α). Dette proteinet er involvert i kroppens inflammatoriske prosesser, og ved å blokkere det er det mulig å redusere betennelse i kroppen.
Leddgikt
Revmatoid artritt er en inflammatorisk leddsykdom. Hvis du har aktiv revmatoid artritt, vil du i utgangspunktet bli behandlet med andre medisiner. Hvis du ikke svarer tilstrekkelig på disse legemidlene, vil du bli behandlet med Simponi i kombinasjon med et annet legemiddel kalt metotreksat for:
- Reduser tegn og symptomer på sykdommen.
- Bremse skaden på bein og ledd.
- Forbedre fysisk funksjon.
Psoriasisartritt
Psoriasisartritt er en inflammatorisk leddsykdom, vanligvis ledsaget av psoriasis, en inflammatorisk hudsykdom. Hvis du har aktiv psoriasisartritt, vil du først bli behandlet med andre medisiner. Hvis du ikke svarer tilstrekkelig på disse legemidlene, blir du behandlet med Simponi for:
- Reduser tegn og symptomer på sykdommen.
- Bremse skaden på bein og ledd.
- Forbedre fysisk funksjon.
Ankyloserende spondylitt og ikke-radiografisk aksial spondylartritt
Ankyloserende spondylitt og ikke-radiografisk aksial spondylartritt er inflammatoriske sykdommer i ryggraden. Hvis du har ankyloserende spondylitt eller ikke-radiografisk aksial spondylartritt, vil du først bli behandlet med andre medisiner. Hvis du ikke svarer tilstrekkelig på disse legemidlene, vil du bli behandlet med Simponi for å:
- Reduser tegn og symptomer på sykdommen.
- Forbedre fysisk funksjon.
Ulcerøs kolitt
Ulcerøs kolitt er en inflammatorisk tarmsykdom. Hvis du har ulcerøs kolitt, vil du bli gitt andre medisiner først. Hvis du ikke svarer tilstrekkelig på disse legemidlene, får du Simponi for å behandle sykdommen din.
Polyartikulær juvenil idiopatisk artritt
Polyartikulær juvenil idiopatisk artritt er en inflammatorisk sykdom som forårsaker smerter og hevelse i leddene hos barn.Hvis barnet ditt har polyartikulær juvenil idiopatisk artritt, vil barnet ditt få andre medisiner først. Hvis barnet ditt ikke svarer tilstrekkelig på disse legemidlene, vil barnet ditt bli gitt Simponi i kombinasjon med metotreksat for å behandle sykdommen.
Kontraindikasjoner Når Simponi ikke skal brukes
Ikke bruk Simponi:
- Hvis du er allergisk (overfølsom) overfor golimumab eller noen av de andre innholdsstoffene i dette legemidlet (listet opp i avsnitt 6).
- Hvis du har tuberkulose (TB) eller annen alvorlig infeksjon.
- Hvis du har moderat eller alvorlig hjertesvikt.
Hvis du er usikker på om noen av de ovennevnte betingelsene gjelder for deg, snakk med legen din, apoteket eller sykepleieren før du bruker Simponi.
Forholdsregler for bruk Hva du må vite før du bruker Simponi
Rådfør deg med lege, apotek eller sykepleier før du bruker Simponi.
Infeksjoner
Fortell legen din umiddelbart hvis du allerede har eller har symptomer på infeksjon under eller etter behandling med Simponi. Symptomer på infeksjonen inkluderer feber, hoste, kortpustethet, influensalignende symptomer, diaré, sår, tannproblemer eller en brennende følelse ved vannlating.
- Du kan lettere få infeksjoner når du bruker Simponi.
- Infeksjoner kan utvikle seg raskere og bli mer alvorlige. Også infeksjoner fra fortiden kan komme tilbake.
Tuberkulose (TB)
Fortell legen din umiddelbart hvis du merker symptomer på TB under behandlingen. Symptomer på TB inkluderer vedvarende hoste, vekttap, tretthet, feber eller nattesvette.
- Noen få tilfeller av TB er rapportert hos pasienter behandlet med Simponi, i sjeldne tilfeller selv hos pasienter som har blitt behandlet med medisiner mot TB. Legen din vil ta tester for å se om du har TB. Legen vil registrere disse testene på pasientkortet.
- Det er veldig viktig at du forteller legen din om du har hatt TB tidligere, eller hvis du har kommet i nær kontakt med noen som har eller har hatt TB.
- Hvis legen din tror du er i fare for tuberkulose, kan du bli behandlet med medisiner mot tuberkulose før du får Simponi.
Hepatitt B -virus (HBV)
- Fortell legen din dersom du er bærer eller har eller har hatt hepatitt B før du får Simponi
- Fortell legen din dersom du tror du kan ha risiko for å pådra deg hepatitt B
- Legen din bør vurdere om du har hepatitt B
- Behandling med TNF -blokkere som Simponi kan føre til at hepatitt B -virus blir reaktivert hos pasienter som bærer dette viruset, noe som i noen tilfeller kan forårsake død.
Invasive soppinfeksjoner
Fortell legen din umiddelbart hvis du har bodd i eller reist til et "område hvor infeksjoner forårsaket av spesifikke sopptyper som kan påvirke lungene eller andre deler av kroppen (kalt histoplasmose, coccidioidomycosis eller blastomycosis) er vanlige. Spør legen din om du vet ikke om disse soppinfeksjonene er vanlige i området der du bodde eller reiste.
Kreft og lymfom
Fortell legen din dersom du har eller har hatt lymfom (en type blodkreft) eller andre typer kreft før du får Simponi.
- Hvis du bruker Simponi eller andre TNF -blokkere, kan du øke risikoen for å utvikle lymfom eller annen type kreft.
- Pasienter med alvorlig revmatoid artritt eller andre inflammatoriske tilstander som har lidd av denne sykdommen i lang tid, kan ha en høyere risiko enn gjennomsnittlig for å utvikle lymfom.
- Kreft, inkludert uvanlige, noen ganger dødelige kreftformer, er rapportert hos barn og ungdomspasienter som bruker TNF-blokkerende legemidler.
- I sjeldne tilfeller har det blitt observert en spesifikk og alvorlig type lymfom kalt hepatosplenisk T-cellelymfom hos pasienter som tar andre TNF-blokkere. De fleste av disse pasientene var ungdom eller unge mannlige voksne. Denne kreftformen har vanligvis resultert i død. Nesten alle disse pasientene fikk også medisiner kjent som azatioprin eller 6-merkaptopurin. Fortell legen din dersom du bruker azatioprin eller 6-merkaptopurin med Simponi.
- Pasienter med alvorlig vedvarende astma, kronisk obstruktiv lungesykdom (KOL) eller storrøykere kan ha økt risiko for kreft ved behandling med Simponi. Hvis du har alvorlig vedvarende astma, KOL eller er storrøyker, bør du diskutere med legen din om behandling med en TNF -blokker er hensiktsmessig.
- Noen golimumab-behandlede pasienter har utviklet visse typer hudkreft. Fortell legen din dersom du opplever noen form for endring i hudens utseende eller vekst på huden under eller etter behandlingen.
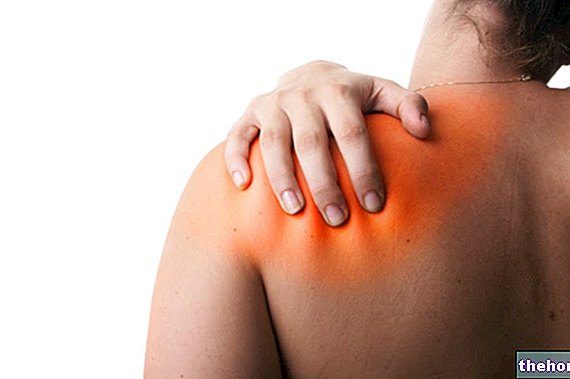
Hjertefeil
Fortell legen din umiddelbart hvis du merker nye eller forverrede symptomer på hjertesvikt. Symptomer på hjertesvikt inkluderer kortpustethet eller hevelse i føttene.
- Tilfeller av ny debut eller forverring av kongestiv hjertesvikt er rapportert med TNF -blokkere, inkludert Simponi. Noen av disse pasientene har dødd.
- Hvis du har mild hjertesvikt og blir behandlet med Simponi, vil legen din følge deg nøye.
Nervesystemet sykdom
Fortell legen din umiddelbart hvis du noen gang har blitt diagnostisert med eller utvikler symptomer på en demyeliniserende sykdom, for eksempel multippel sklerose. Symptomer kan omfatte endringer i syn, svakhet i armer og ben, nummenhet eller prikking i noen deler av kroppen. Legen din vil avgjøre om du skal ta Simponi.
Tannoperasjoner eller prosedyrer
- Fortell legen din dersom du skal utføre tannoperasjoner eller prosedyrer.
- Fortell kirurgen eller tannlegen som utfører prosedyren at du blir behandlet med Simponi ved å vise pasientkortet.
Autoimmune sykdommer
Fortell legen din dersom du utvikler symptomer på en sykdom som kalles lupus. Symptomer inkluderer vedvarende utslett, feber, leddsmerter og tretthet.
- I sjeldne tilfeller har mennesker behandlet med TNF -blokkere utviklet lupus.
Sykdommer i blodet
Hos noen pasienter kan det hende at kroppen ikke produserer nok blodceller som hjelper kroppen med å bekjempe infeksjoner eller hjelpe til med å stoppe blødninger. Hvis du har vedvarende feber som du ikke forstår, lett får blåmerker eller blør eller ser blek ut, må du kontakte legen din umiddelbart. Legen din kan beslutte å stoppe behandlingen.
Hvis du ikke er sikker på om noen av de ovennevnte betingelsene gjelder for deg, snakk med legen din eller apoteket før du bruker Simponi.
Vaksinasjoner
Fortell legen din dersom du nylig har blitt vaksinert eller planlegger å bli vaksinert.
- Du må ikke motta visse (levende) vaksiner mens du blir behandlet med Simponi.
- Noen vaksinasjoner kan forårsake infeksjoner. Hvis du mottok Simponi mens du var gravid, kan babyen din ha en økt risiko for å få denne infeksjonen i omtrent seks måneder etter den siste dosen som ble mottatt under graviditet.Det er viktig å fortelle barnelege og annet helsepersonell om bruk av Simponi. kan bestemme når barnet skal få noen vaksine.
Snakk med barnets lege angående vaksinasjoner for barnet ditt. Hvis det er mulig, bør barnet ditt være oppdatert med alle vaksinasjoner før du bruker Simponi.
Smittsomme terapeutiske midler
Snakk med legen din dersom du nylig har tatt eller planlegger å ta behandling med et smittsomt terapeutisk middel (for eksempel BCG -instillasjon som brukes til å behandle kreft).
Allergiske reaksjoner
Fortell legen din umiddelbart hvis du får symptomer på en allergisk reaksjon etter behandling med Simponi. Symptomer på en allergisk reaksjon kan omfatte hevelse i ansikt, lepper, munn eller svelg som kan forårsake svelging eller pustevansker, utslett, elveblest, hevelse i hender, føtter og ankler.
- Noen av disse reaksjonene kan være alvorlige eller sjelden livstruende.
- Noen av disse reaksjonene oppstår etter den første administreringen av Simponi.
Barn og ungdom
Simponi anbefales ikke for barn som veier mindre enn 40 kg med polyartikulær juvenil idiopatisk artritt eller hos barn og ungdom under 18 år for andre tilstander.
Interaksjoner Hvilke medisiner eller matvarer kan endre effekten av Simponi
- Fortell legen din eller apoteket dersom du bruker, nylig har brukt eller planlegger å bruke andre legemidler, inkludert andre legemidler for behandling av revmatoid artritt, polyartikulær juvenil idiopatisk artritt, psoriasisartritt, ankyloserende spondylitt, aksial spondyloartritt, ikke-radiografisk eller ulcerøs kolitt.
- Du bør ikke ta Simponi med medisiner som inneholder virkestoffet anakinra eller abatacept. Disse medisinene brukes til behandling av revmatiske sykdommer.
- Fortell legen din eller apoteket dersom du bruker andre legemidler som påvirker immunsystemet.
- Det kan ikke behandles med visse (levende) vaksiner mens du bruker Simponi.
Hvis du er usikker på om noen av de ovennevnte betingelsene gjelder for deg, snakk med legen din eller apoteket før du bruker Simponi.
Advarsler Det er viktig å vite at:
Graviditet og amming
Snakk med legen din før du bruker Simponi hvis:
- Du er gravid eller planlegger å bli gravid mens du bruker Simponi. Effekten av dette legemidlet hos gravide er ikke kjent. Det anbefales ikke å bruke Simponi til gravide. Hvis du skal behandles med Simponi, bør du unngå å bli gravid ved å bruke tilstrekkelig prevensjon under behandlingen og i minst 6 måneder etter din siste Simponi -injeksjon.
- Før amming må siste Simponi -behandling være minst 6 måneder tidligere Du må slutte å amme hvis Simponi skal gis deg.
- Hvis du mottok Simponi under graviditeten, kan babyen din ha økt risiko for å få en infeksjon. Det er viktig å fortelle barnelege og annet helsepersonell om din bruk av Simponi før babyen din får noen vaksiner (for mer informasjon, se avsnittet på vaksinasjoner).
Rådfør deg med lege eller apotek før du bruker dette legemidlet hvis du er gravid eller ammer, tror at du kan være gravid eller planlegger å bli gravid.
Kjøring og bruk av maskiner
Simponi kan svekke evnen til å kjøre bil og bruke verktøy eller maskiner litt. Du kan føle deg svimmel etter å ha brukt Simponi. I så fall må du ikke kjøre bil eller bruke verktøy eller maskiner.
Simponi inneholder latex og sorbitol
Følsomhet for latex
Latexfølsomhet En del av den ferdigfylte pennen, hetten som dekker nålen, inneholder latex Siden latex kan forårsake alvorlige allergiske reaksjoner, må du fortelle legen din før du bruker Simponi hvis du eller omsorgspersonen din er allergisk mot latex.
Sorbitol intoleranse
Simponi inneholder sorbitol (E420). Hvis legen din har fortalt deg at du ikke tåler noen sukkerarter, må du kontakte legen din før du tar denne medisinen.
Dosering og bruksmåte Hvordan bruke Simponi: Dosering
Bruk alltid dette legemidlet nøyaktig slik legen din eller apoteket har fortalt deg. Hvis du er i tvil, bør du kontakte lege eller apotek.
Hvor mye Simponi gis
Revmatoid artritt, psoriasisartritt og aksial spondylartritt, inkludert ankyloserende spondylitt og ikke-radiografisk aksial spondylartritt:
- Den anbefalte dosen er 50 mg (innholdet i 1 ferdigfylt penn) administrert en gang i måneden, samme dag hver måned.
- Snakk med legen din før du tar den fjerde dosen. Legen din vil avgjøre om du skal fortsette Simponi -behandlingen.
- Hvis du veier mer enn 100 kg, kan dosen økes til 100 mg (innholdet i 2 ferdigfylte penner), gitt en gang i måneden, alltid på samme dag hver måned.
Polyartikulær ungdomsidiopatisk artritt:
- Den anbefalte dosen er 50 mg gitt en gang i måneden, samme dag hver måned.
- Snakk med barnets lege før barnet ditt tar den fjerde dosen. Barnas lege vil avgjøre om du skal fortsette Simponi -behandlingen.
Ulcerøs kolitt
- Tabellen nedenfor viser hvordan du generelt vil bruke dette legemidlet.
- Hos pasienter som veier mindre enn 80 kg, 50 mg (innholdet i 1 ferdigfylt penn) 4 uker etter siste behandling, deretter hver fjerde uke deretter.
- Hos pasienter som veier 80 kg eller mer, 100 mg (innholdet i 2 ferdigfylte penner) 4 uker etter siste behandling, deretter hver 4. uke.
Hvordan Simponi gis
- Simponi gis ved injeksjon under huden (subkutant).
- Først vil legen din eller pleiepersonalet injisere Simponi, men du og legen din kan bestemme at du kan injisere Simponi selv.I dette tilfellet vil du bli instruert i hvordan du skal injisere Simponi selv.
Snakk med legen din dersom du har spørsmål om selvadministrering av en injeksjon. På slutten av dette pakningsvedlegget finner du detaljerte "Instruksjoner for administrering".
Dersom du har glemt å bruke Simponi
Hvis du glemmer å bruke Simponi på den planlagte dagen, injiser den glemte dosen så snart du husker det.
Ikke bruk en dobbel dose for å gjøre opp for en glemt dose.
Når skal neste dose injiseres:
- Hvis du er mindre enn 2 uker forsinket, injiser den glemte dosen så snart du husker det og fortsett å følge den opprinnelige timeplanen.
- Hvis du er mer enn 2 uker forsinket, injiser den glemte dosen så snart du husker det og fortell legen din eller apoteket og spør når du skal ta din neste dose.
Spør legen din, apoteket eller sykepleieren hvis du ikke er sikker på hva du skal gjøre.
Hvis du slutter å bruke Simponi
Hvis du vurderer å stoppe Simponi, snakk med legen din eller apoteket først.
Spør lege eller apotek hvis du har ytterligere spørsmål om bruken av dette legemidlet.
Overdosering Hva du skal gjøre hvis du har tatt for mye Simponi
Hvis du har brukt eller gitt for mye Simponi (injisert for mye i en enkelt dose eller brukt det for ofte), må du fortelle det til legen din eller apoteket umiddelbart. Ta alltid med deg ytterkartongen og dette pakningsvedlegget, selv om det er tomt.
Bivirkninger Hva er bivirkningene av Simponi
Som alle andre legemidler kan dette legemidlet forårsake bivirkninger, men ikke alle får det. Noen pasienter kan oppleve alvorlige bivirkninger som kan kreve medisinsk behandling.Risikoen for noen bivirkninger er høyere med dosen på 100 mg sammenlignet med dosen på 50 mg. Bivirkninger kan også oppstå flere måneder etter siste injeksjon.
Fortell legen din umiddelbart hvis du merker noen av følgende alvorlige bivirkninger av Simponi, som inkluderer:
- allergiske reaksjoner som kan være alvorlige eller sjelden livstruende (sjeldne). Symptomer på en allergisk reaksjon kan omfatte hevelse i ansikt, lepper, munn eller svelg som kan forårsake svelging eller pustevansker, utslett, elveblest, hevelse i hender, føtter eller ankler. Noen av disse reaksjonene skjedde etter den første administreringen av Simponi.
- alvorlige infeksjoner (inkludert TB, bakterielle infeksjoner inkludert alvorlige blodinfeksjoner og lungebetennelse, alvorlige soppinfeksjoner og andre opportunistiske infeksjoner) (vanlig). Symptomer på en infeksjon kan omfatte feber, tretthet, (vedvarende) hoste, kortpustethet, influensalignende symptomer, vekttap, nattesvette, diaré, sår, tannproblemer og en brennende følelse ved vannlating.
- reaktivering av hepatitt B -viruset hvis du er bærer eller tidligere har hatt hepatitt B (sjelden). Symptomene kan være gulfarging av hud og øyne, mørk brun urin, smerter i høyre side av magen, feber, ubehag, ubehag og trøtthet.
- nervesystemet sykdom som multippel sklerose (sjelden). Symptomer på sykdom i nervesystemet kan omfatte endringer i syn, svakhet i armer eller ben, nummenhet eller prikking i noen deler av kroppen.
- kreft i lymfeknuter (lymfom) (sjelden). Symptomer på lymfom kan omfatte hovne lymfeknuter, vekttap eller feber.
- hjertesvikt (sjelden). Symptomer på hjertesvikt kan være kortpustethet eller hevelse i føttene.
- tegn på lidelser i immunsystemet kalt: - lupus (sjelden). Symptomer kan omfatte leddsmerter eller utslett på kinnene eller armene som er følsom for solen. - sarkoidose (sjelden). Symptomene kan være vedvarende hoste, kortpustethet, brystsmerter, feber, hovne lymfeknuter, vekttap, utslett og tåkesyn.
- hevelse i små blodkar (vaskulitt) (sjelden). Symptomer kan omfatte feber, hodepine, vekttap, nattesvette, utslett og nerveproblemer som nummenhet og prikking.
- hudkreft (uvanlig). Hudkreft symptomer kan omfatte endringer i hudens utseende eller vekst på huden.
- blodsykdom (vanlig). Symptomer på blodsykdom kan omfatte feber som ikke forsvinner, sterk tendens til blåmerker eller blødninger eller et veldig blekt utseende.
- blodkreft (leukemi) (sjelden). Symptomer på leukemi kan omfatte feber, tretthet, hyppige infeksjoner, blåmerker og nattesvette.
Fortell legen din umiddelbart hvis du merker noen av symptomene ovenfor.
Følgende ekstra bivirkninger er observert med Simponi:
Svært vanlige bivirkninger (kan forekomme hos flere enn 1 av 10 personer):
- Øvre luftveisinfeksjoner, ondt i halsen eller heshet, forkjølelse
Vanlige bivirkninger (kan forekomme hos opptil 1 av 10 personer):
- Unormale levertester (økte leverenzymer), funnet under blodprøver utført av legen din
- Føler meg svimmel
- Hodepine
- Følelse av nummenhet eller prikking
- Overfladiske soppinfeksjoner
- Abscess
- Bakterielle infeksjoner (for eksempel cellulitt)
- Reduksjon av røde blodlegemer
- Positiv blodprøve for lupus
- Allergiske reaksjoner
- Fordøyelsesbesvær
- Magesmerter
- Føler meg kvalm
- Innflytelse
- Bronkitt
- Bihulebetennelse
- Ansiktsherpes
- Høyt blodtrykk
- Feber
- Astma, kortpustethet, tungpustethet
- Forstyrrelser i mage og tarm som inkluderer betennelse i det indre av mage og tykktarm som kan forårsake feber
- Smerter og sår i munnen
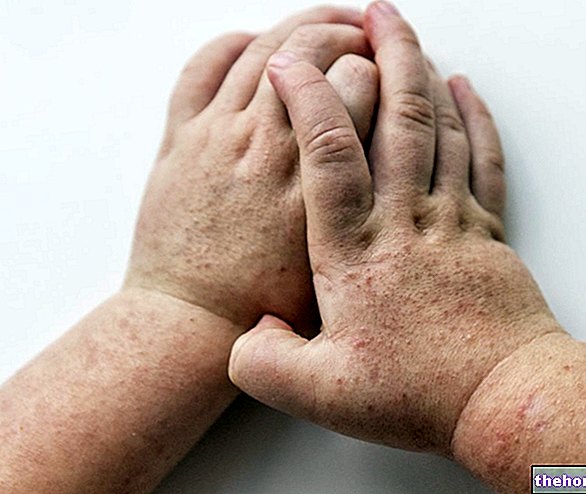
- Reaksjoner på injeksjonsstedet (inkludert rødhet, hardhet, smerte, blåmerker, kløe, prikking og irritasjon)
- Hårtap
- Utslett og kløende hud
- Søvnvansker
- Depresjon
- Følelse av svakhet
- Brudd i beinene
- Brystsmerter
Mindre vanlige bivirkninger (kan forekomme hos opptil 1 av 100 personer):
- Nyreinfeksjon
- Kreft, inkludert hudkreft og ikke-kreftige klumper eller små masser, inkludert føflekker
- Hudblemmer
- Psoriasis (inkludert håndflatene og / eller fotsålene og / eller i form av hudpustler)
- Reduksjon av blodplater
- Reduksjon av hvite blodlegemer
- Kombinert reduksjon av blodplater, røde blodlegemer og hvite blodlegemer
- Skjoldbrusk lidelser
- Økning i blodsukkernivået
- Økning i kolesterolnivået i blodet
- Balanseforstyrrelser
- Synsforstyrrelser
- Følelse av uregelmessig hjerterytme
- Innsnevring av blodkar i hjertet
- Blodpropp
- Rødhet
- Forstoppelse
- Kronisk betennelse i lungene
- Sure oppstøt
- Steiner i gallen
- Leverproblemer
- Brystsykdommer
- Menstruasjonsforstyrrelser
Sjeldne bivirkninger (kan forekomme hos opptil 1 av 1000 personer):
- Benmargs manglende evne til å produsere blodceller
- Infeksjon av ledd eller omkringliggende vev
- Vanskelig helbredelse
- Betennelse i blodårene i de indre organene
- Leukemi
- Melanom (en type hudkreft)
- Skallende hud
- Immunologiske lidelser som kan påvirke lungene, huden og lymfeknuter (svært vanlig som sarkoidose)
- Smerter og misfarging i fingre eller tær
- Smaksforstyrrelser
- Blæreforstyrrelser
- Nyreproblemer
- Betennelse i blodårene i huden forårsaker utslett
Bivirkninger med frekvens ikke kjent:
- Merkelcellekarsinom (en type hudkreft)
- En sjelden blodkreft som hovedsakelig rammer unge mennesker (hepatosplenisk T-cellelymfom)
Rapportering av bivirkninger
Hvis du får bivirkninger, snakk med legen din, apoteket eller sykepleieren. Dette inkluderer mulige bivirkninger som ikke er nevnt i dette pakningsvedlegget. Du kan også rapportere bivirkninger direkte via det nasjonale meldingssystemet som er oppført i vedlegg V. Du kan hjelpe gi mer informasjon om sikkerheten til dette legemidlet.
Utløp og oppbevaring
- Hold denne medisinen utilgjengelig for barn.
- Ikke bruk dette legemidlet etter utløpsdatoen som er angitt på etiketten og esken etter "Utløpsdato". Utløpsdatoen refererer til den siste dagen i den måneden.
- Oppbevares i kjøleskap (2 ° C-8 ° C). Ikke frys.
- Oppbevar den ferdigfylte pennen i ytterkartongen for å beskytte medisinen mot lys.
- Ikke bruk dette legemidlet hvis du merker at væsken ikke er lys til lysegul, er grumsete eller inneholder fremmede partikler.
- Ikke kast medisiner i avløpsvann eller husholdningsavfall. Spør legen din eller apoteket om hvordan du skal kaste medisiner du ikke bruker lenger. Dette vil bidra til å beskytte miljøet.
Annen informasjon
Hva Simponi inneholder
Den aktive ingrediensen er golimumab. En 0,5 ml ferdigfylt penn inneholder 50 mg golimumab.
Andre innholdsstoffer er sorbitol (E420), L-histidin, L-histidinmonohydrokloridmonohydrat, polysorbat 80 og vann til injeksjonsvæsker.
Simponi ser ut og innholdet i pakningen
Simponi leveres som en injeksjonsvæske, oppløsning i en ferdigfylt penn til engangsbruk. Simponi er tilgjengelig i pakninger som inneholder 1 ferdigfylt penn og flerpakninger som inneholder 3 (3 pakninger med 1) ferdigfylte penner. Det er ikke sikkert at alle pakningsstørrelser blir markedsført.
Løsningen er klar til lett opaliserende (skinnende som en perle), fargeløs til lysegul og kan inneholde noen små gjennomsiktige eller hvite proteinpartikler. Ikke bruk Simponi hvis løsningen har endret farge, er grumsete eller inneholder synlige fremmede partikler.
Kildepakningsvedlegg: AIFA (Italian Medicines Agency). Innhold publisert i januar 2016. Informasjonen som er tilstede er kanskje ikke oppdatert.
For å få tilgang til den mest oppdaterte versjonen, er det lurt å gå til nettstedet til AIFA (Italian Medicines Agency). Ansvarsfraskrivelse og nyttig informasjon.
01.0 LEGEMIDLETS NAVN
SIMPONI 50 MG INJEKTERbar LØSNING
02.0 KVALITATIV OG KVANTITATIV SAMMENSETNING
Simponi 50 mg injeksjonsvæske, oppløsning i ferdigfylt penn
En 0,5 ml ferdigfylt penn inneholder 50 mg golimumab *.
Simponi 50 mg injeksjonsvæske, oppløsning i ferdigfylt sprøyte
En 0,5 ml ferdigfylt sprøyte inneholder 50 mg golimumab *.
* IgG1 humant monoklonalt antistoff? produsert fra en murin hybridomcellelinje med rekombinant DNA -teknologi.
Hjelpestoff med kjent effekt:
Hver ferdigfylte penn inneholder: 20,5 mg sorbitol for en dose på 50 mg.
Hver ferdigfylte sprøyte inneholder: 20,5 mg sorbitol for en dose på 50 mg.
For fullstendig liste over hjelpestoffer, se pkt.6.1.
03.0 LEGEMIDDELFORM
Injeksjonsvæske, oppløsning i ferdigfylt penn (injeksjon), SmartJect
Injeksjonsvæske, oppløsning i ferdigfylt sprøyte (injeksjon)
Løsningen er klar til svakt opaliserende, fargeløs til lysegul.
04.0 KLINISK INFORMASJON
04.1 Terapeutiske indikasjoner
Revmatoid artritt (RA)
Simponi, i kombinasjon med metotreksat (MTX), er indikert for:
• behandling av moderat til alvorlig aktiv revmatoid artritt hos voksne pasienter når responsen på sykdomsmodifiserende anti-revmatiske legemidler (DMARD), inkludert MTX, har vært utilstrekkelig.
• behandling av alvorlig, aktiv og progressiv revmatoid artritt hos voksne som ikke tidligere har blitt behandlet med MTX.
Simponi, i kombinasjon med MTX, har vist seg å redusere utviklingen av leddskader målt ved røntgenstråler og forbedre fysisk funksjon.
Ung idiopatisk artritt
Polyartikulær juvenil idiopatisk artritt (PIA)
Simponi i kombinasjon med MTX er indisert for behandling av polyartikulær juvenil idiopatisk artritt hos barn som veier minst 40 kg og som har svart utilstrekkelig på tidligere MTX -behandling.
Psoriasisartritt (AP)
Simponi, alene eller i kombinasjon med metotreksat (MTX), er indisert for behandling av aktiv og progressiv psoriasisartritt hos voksne når responsen på tidligere DMARD -behandlinger har vært utilstrekkelig. Simponi har vist seg å redusere progresjonshastigheten til legemidlet. perifer leddskade, målt ved røntgenstråler hos pasienter med symmetriske polyartikulære sykdomstyper (se pkt. 5.1) og for å forbedre fysisk funksjon.
Aksial spondylartritt
Ankyloserende spondylitt (AS)
Simponi er indisert for behandling av alvorlig, aktiv ankyloserende spondylitt hos voksne som ikke har svart tilstrekkelig på konvensjonelle behandlinger.
Ikke-radiografisk aksial spondylartritt (aksial SpA nr)
Simponi er indisert for behandling av voksne pasienter med alvorlig aktiv ikke-radiografisk aksial spondylartritt med objektive tegn på betennelse som indikert av forhøyet C-reaktivt protein (CRP) og / eller magnetisk resonansavbildning (MR) som har hatt utilstrekkelig respons. eller er intolerante overfor ikke-steroide antiinflammatoriske legemidler (NSAIDs).
Ulcerøs kolitt (CU)
Simponi er indisert for behandling av moderat til alvorlig aktiv ulcerøs kolitt hos voksne pasienter som ikke har svart tilstrekkelig på konvensjonell behandling, inkludert kortikosteroider og 6-merkaptopurin (6-MP) eller azatioprin (AZA), eller som er intolerante eller som det er en medisinsk kontraindikasjon for disse behandlingene.
04.2 Dosering og administrasjonsmåte
Simponi-behandling bør initieres og overvåkes av spesialister som har erfaring med diagnostisering og behandling av revmatoid artritt, polyartikulær juvenil idiopatisk artritt, psoriasisartritt, ankyloserende spondylitt, ikke-radiografisk aksial spondyloartritt eller ulcerøs kolitt. Pasienter behandlet med Simponi bør gis pasientvarsel. Kort.
Dosering
Leddgikt
Simponi 50 mg gis en gang i måneden, samme dag hver måned.
Simponi må administreres samtidig med MTX.
Psoriasisartritt, ankyloserende spondylitt eller ikke-radiografisk aksial spondylartritt
Simponi 50 mg gis en gang i måneden, samme dag hver måned.
For alle indikasjonene ovenfor tyder tilgjengelige data på at klinisk respons vanligvis oppnås innen 12-14 uker etter oppstart av behandlingen (etter 3-4 doser) Det bør vurderes å fortsette behandlingen hos pasienter som ikke viser tegn på terapeutisk fordel innenfor denne tidsrammen.
Pasienter med en kroppsvekt større enn 100 kg
For alle de ovennevnte indikasjonene, hos pasienter med RA, AP, SA eller aksial SpA nr som veier mer enn 100 kg, som ikke oppnår tilstrekkelig klinisk respons etter 3 eller 4 doser, øker dosen golimumab opp til 100 mg en gang i måneden, med tanke på den økte risikoen for noen alvorlige bivirkninger med 100 mg dosen sammenlignet med dosen på 50 mg (se pkt. 4.8) Det bør vurderes å fortsette behandlingen hos pasienter som ikke viser tegn på terapeutisk fordel etter å ha mottatt 3 4 tilleggsdoser på 100 mg.
Ulcerøs kolitt
Pasienter med en kroppsvekt på mindre enn 80 kg
Simponi gitt som en startdose på 200 mg, etterfulgt av 100 mg i uke 2, deretter 50 mg hver fjerde uke deretter (se pkt.5.1).
Pasienter med en kroppsvekt større enn eller lik 80 kg
Simponi gitt som en startdose på 200 mg, etterfulgt av 100 mg i uke 2, deretter 100 mg hver fjerde uke deretter (se pkt.5.1).
Under vedlikeholdsbehandling kan kortikosteroider gradvis reduseres i henhold til retningslinjer for klinisk praksis.
Tilgjengelige data tyder på at klinisk respons vanligvis oppnås innen 12-14 uker etter behandling (etter 4 doser). Det bør vurderes å fortsette behandlingen hos pasienter som ikke viser tegn på terapeutisk nytte innen denne tidsperioden.
Glemt dose
Hvis en pasient glemmer å injisere Simponi på den planlagte dagen, bør den glemte dosen injiseres så snart pasienten husker det. Pasienter bør instrueres til ikke å injisere en dobbel dose for å gjøre opp for en glemt dose.
Den neste dosen bør administreres i henhold til følgende veiledning:
• Hvis forsinket dose er mindre enn 2 uker, bør pasienten injisere den glemte dosen og fortsette å følge den opprinnelige planen.
• Hvis doseringsforsinkelsen er mer enn 2 uker, bør pasienten injisere den glemte dosen, og en ny doseringsplan må defineres fra datoen for denne injeksjonen.
Spesielle populasjoner
Eldre (≥ 65 år gammel)
Ingen dosejustering er nødvendig hos eldre.
Nedsatt nyre- og leverfunksjon
Simponi har ikke blitt studert i disse pasientpopulasjonene. Ingen doseanbefalinger kan gis.
Pediatrisk populasjon
Sikkerhet og effekt av Simponi hos pasienter under 18 år for andre indikasjoner enn pIA er ikke fastslått.
Polyartikulær juvenil idiopatisk artritt
Simponi 50 mg gis en gang i måneden, samme dag i hver måned, for barn med en kroppsvekt på minst 40 kg.
Tilgjengelige data tyder på at klinisk respons vanligvis oppnås innen 12-14 uker etter behandling (etter 3-4 doser). Fortsettelse av behandlingen bør vurderes på nytt hos barn som ikke viser tegn på terapeutisk nytte innen denne tidsperioden.
Administrasjonsmåte
Simponi er til subkutan bruk. Etter tilstrekkelig opplæring i den subkutane injeksjonsteknikken, vil pasientene kunne injisere Simponi selv hvis legen deres finner det mulig, med passende medisinsk tilsyn om nødvendig. Pasienter bør instrueres til å injisere hele Simponi -mengden i samsvar med de fullstendige administrasjonsinstruksjonene i pakningsvedlegget. Hvis det er nødvendig med flere injeksjoner, bør injeksjonene administreres på forskjellige kroppssteder.
For instruksjoner om administrasjon, se avsnitt 6.6.
04.3 Kontraindikasjoner
Overfølsomhet overfor virkestoffet eller overfor noen av hjelpestoffene listet opp i pkt.6.1.
Aktiv tuberkulose (TB) eller andre alvorlige infeksjoner som sepsis og opportunistiske infeksjoner (se pkt. 4.4).
Pasienter med moderat til alvorlig hjertesvikt (NYHA - New York Heart Association Class III / IV) (se pkt. 4.4).
04.4 Spesielle advarsler og passende forholdsregler for bruk
Infeksjoner
Før, under og etter Simponi -behandling, bør pasientene overvåkes nøye for infeksjoner inkludert tuberkulose. Siden eliminering av golimumab kan ta opptil 5 måneder, bør overvåking fortsette i løpet av denne perioden. Videre behandling med Simponi bør ikke gis hvis en pasient utvikler alvorlige infeksjoner eller sepsis (se pkt. 4.3).
Simponi må ikke brukes til pasienter med klinisk signifikant, aktiv infeksjon. Forsiktighet er nødvendig når man vurderer bruk av Simponi hos pasienter med kronisk infeksjon eller en historie med tilbakevendende infeksjoner. Pasienter bør informeres på passende måte om behovet for å unngå eksponering for potensielle risikofaktorer for infeksjoner.
Pasienter som tar TNF-blokkerende medisiner er mer utsatt for alvorlige infeksjoner.
Hos pasienter behandlet med Simponi er det rapportert om bakterielle infeksjoner (inkludert sepsis og lungebetennelse), mykobakteriell (inkludert TB), invasive soppinfeksjoner og opportunistiske infeksjoner, inkludert de med dødelig utgang. Noen av disse alvorlige infeksjonene har utviklet seg hos pasienter som samtidig får immunsuppressiv behandling, som i tillegg til den underliggende sykdommen kan disponere dem for infeksjoner. Pasienter som utvikler en ny infeksjon under behandling med Simponi, bør overvåkes nøye og gjennomgå en "grundig diagnostisk evaluering. Simponi -administrering bør avbrytes hvis en pasient utvikler en ny alvorlig infeksjon eller sepsis og passende" initiering. Antimikrobiell eller soppdrepende behandling til infeksjonen er For pasienter som har bodd i eller reist til områder der invasive soppinfeksjoner som histoplasmose, koksidioidomykose eller blastomykose er endemiske, bør fordeler og risiko ved Simponi-behandling vurderes nøye før behandling med Simponi påbegynnes. Hos høyrisikopasienter behandlet med Simponi, en invasiv soppinfeksjon bør mistenkes hvis de utvikler alvorlig systemisk sykdom. Om mulig bør diagnosen og administrasjonen av empirisk soppdrepende behandling hos disse pasientene gjøres i samråd med en lege med erfaring i behandling av pasienter med invasive soppinfeksjoner.
Tuberkulose
Tilfeller av tuberkulose er rapportert hos pasienter behandlet med Simponi. Det skal bemerkes at det i de fleste tilfellene var ekstrapulmonal tuberkulose, både lokalisert og diffus.
Før du starter Simponi -behandling, bør alle pasientene evalueres for både aktiv og inaktiv ("latent") tuberkulose. Denne evalueringen bør inneholde en detaljert medisinsk historie inkludert en personlig historie med tuberkulose eller mulig tidligere kontakt med en kilde til TB -infeksjon og tidligere og / eller samtidig immunsuppressive behandlinger. Passende diagnostiske tester som hud- eller blodtuberkulinprøver og røntgenstråler bør utføres hos alle pasienter (lokale retningslinjer kan gjelde). Det anbefales at disse testene rapporteres på pasientkortet. Foreskrivere blir minnet om risikoen for falske negative tuberkulinhudtestresultater, spesielt hos alvorlig syke eller immunsvekkede pasienter.
Hvis aktiv tuberkulose er diagnostisert, må Simponi -behandling ikke startes (se pkt. 4.3).
Hvis det mistenkes latent tuberkulose, bør en lege med erfaring i behandling av tuberkulose konsulteres. I alle situasjonene beskrevet nedenfor må nytte / risiko -balansen for Simponi -behandlingen vurderes nøye.
Hvis det er påvist inaktiv ("latent") tuberkulose, bør antituberkulosebehandling for latent tuberkulose startes før Simponi -behandlingen startes, i henhold til lokale retningslinjer.
Hos pasienter som har mange eller betydelige risikofaktorer for tuberkulose og har en negativ test for latent tuberkulose, bør behandling mot tuberkulose vurderes før oppstart av Simponi.Bruk av anti-tuberkuloseterapi bør også vurderes før oppstart av Simponi-behandling hos pasienter som tidligere har hatt latent eller aktiv tuberkulose og som ikke har et tilstrekkelig behandlingsforløp.
Tilfeller av aktiv tuberkulose har oppstått hos pasienter behandlet med Simponi under og etter behandling for latent tuberkulose. Pasienter behandlet med Simponi bør overvåkes nøye for tegn og symptomer på aktiv tuberkulose, inkludert pasienter som har testet negativt for latent tuberkulose, pasienter som blir behandlet for latent tuberkulose, eller pasienter som tidligere har blitt behandlet for latent tuberkulose. "Tuberkuloseinfeksjon.
Alle pasienter bør rådes til å oppsøke lege hvis tegn / symptomer som tyder på tuberkulose (f.eks. Vedvarende hoste, sløsing / vekttap, lav feber) oppstår under eller etter behandling med Simponi.
Reaktivering av hepatitt B -virus
Reaktivering av hepatitt B har blitt observert hos pasienter behandlet med en TNF-antagonist, inkludert Simponi, og som var kroniske bærere av dette viruset (dvs. positivt for overflateantigen). I noen tilfeller har dødelige utfall oppstått.
Pasienter bør evalueres for HBV -infeksjon før behandling med Simponi startes. For pasienter som tester positivt for HBV -infeksjon, anbefales konsultasjon med lege med erfaring i behandling av hepatitt B.
Bærere av hepatitt B -virus som krever Simponi -behandling, bør overvåkes nøye for tegn og symptomer på aktiv hepatitt B -virusinfeksjon i løpet av behandlingen og i mange måneder etter avsluttet behandling. Tilstrekkelige data er tilgjengelige om pasienter med hepatitt B -virus behandlet med antiviral behandling i kombinasjon med TNF-antagonistbehandling for å forhindre reaktivering av hepatitt B. Viruset. Hos pasienter som utvikler hepatitt B-virusreaktivering, bør behandling med Simponi avbrytes og effektiv antiviral behandling med passende støttende behandling startes.
Ondartede neoplasmer og lymfoproliferative sykdommer
Den potensielle rollen til TNF -hemmerterapi i utviklingen av maligniteter er ukjent. Basert på gjeldende kunnskap kan den mulige risikoen for å utvikle lymfom, leukemi eller andre maligniteter hos pasienter behandlet med en TNF -antagonist ikke utelukkes. Det bør utvises forsiktighet ved behandling av TNF -hemmerbehandling hos pasienter med malignitet i historien eller ved videre behandling hos pasienter som utvikler malignitet.
Pediatriske maligne neoplasmer
Etter markedsføring er det rapportert om maligniteter, noen dødelige, blant barn, ungdom og unge voksne (opptil 22 år) behandlet med TNF-blokkerende midler (oppstart av behandling ≤ 18 år. Omtrent halvparten av tilfellene var De andre tilfellene var representert av en rekke forskjellige maligniteter og inkluderte sjeldne maligniteter som vanligvis var forbundet med immunsuppresjon. En risiko for utvikling av malignitet hos barn og ungdom behandlet med TNF -hemmere kan ikke utelukkes.
Lymfom og leukemi
I kontrollerte faser av kliniske studier med alle TNF-hemmermedisiner, inkludert Simponi, ble det observert flere tilfeller av lymfom blant pasienter som fikk anti-TNF-behandling sammenlignet med kontrollpasienter. Under fase IIb og III kliniske studier av Simoni med RA, AP og SA var forekomsten av lymfom hos Simponi-behandlede pasienter høyere enn forventet i befolkningen generelt. Tilfeller av leukemi er rapportert hos Simponi-behandlede pasienter. Det er en økt bakgrunnsrisiko for lymfom og leukemi hos pasienter med revmatoid artritt med langvarig, svært aktiv inflammatorisk sykdom, noe som kompliserer risikostimering.
Sjeldne tilfeller av hepatosplenisk T-cellelymfom (HSTCL) er rapportert etter markedsføring hos pasienter behandlet med andre TNF-blokkerende midler (se pkt. 4.8) Denne sjeldne formen for T-cellelymfom har et ekstremt aggressivt forløp og vanligvis dødelig utfall De fleste tilfeller har forekommet hos unge og unge voksne menn som nesten alle får samtidig behandling med azatioprin (AZA) eller 6-merkaptopurin (6-MP) for inflammatorisk tarmsykdom. Den potensielle risikoen for kombinasjonen av AZA eller 6-MP og Simponi må vurderes nøye.Risiko for utvikling av hepatosplenisk T-cellelymfom hos pasienter behandlet med TNF-blokkerende midler kan ikke utelukkes.
Andre ondartede neoplasmer enn lymfom
I de kontrollerte fasene av fase IIb og III kliniske studier utført med Simponi i RA, AP, SA og CU, var forekomsten av andre maligniteter enn lymfom (unntatt ikke-melanom hudkreft) lik mellom behandlingsgruppen med Simponi og kontrollen .
Tykktarmsdysplasi / karsinom
Det er ikke kjent om golimumab -behandling påvirker risikoen for å utvikle dysplasi eller tykktarmskreft. Alle pasienter med ulcerøs kolitt som har økt risiko for å utvikle tykktarmsdysplasi eller karsinom (for eksempel pasienter med langvarig ulcerøs kolitt eller primær skleroserende kolangitt) eller som har en medisinsk historie med dysplasi eller tykktarmskreft, bør undersøkes for denne dysplasien kl. regelmessige intervaller før du starter behandlingen og i løpet av sykdommen. Denne evalueringen bør omfatte koloskopi og biopsier i samsvar med lokale anbefalinger. Hos pasienter med nydiagnostisert dysplasi som behandles med Simponi, bør risiko / nytte -forholdet hos den enkelte pasient vurderes nøye og om behandlingen skal fortsette.
I en utforskende klinisk studie som evaluerte bruken av Simponi hos pasienter med alvorlig vedvarende astma, ble det rapportert flere tilfeller av malignitet hos Simponi-behandlede pasienter enn hos kontrollpasienter (se pkt. 4.8). Betydningen av disse funnene er ikke kjent.
I en eksplorativ klinisk studie som evaluerte bruken av et annet anti-TNF-middel, infliximab, hos pasienter med moderat til alvorlig kronisk obstruktiv lungesykdom (KOL), ble det rapportert om flere tilfeller av ondartede neoplasmer. Hos pasienter behandlet med infliximab sammenlignet med pasienter i kontrollen Alle pasientene var røykere i lang tid. Derfor bør det utvises forsiktighet ved evaluering av bruk av en TNF -antagonist hos KOL -pasienter, så vel som hos pasienter med økt risiko for malignitet som storrøykere.
Hudsvulster
Melanom og Merkel cellekarsinom er rapportert hos pasienter behandlet med TNF-hemmende midler, inkludert Simponi (se pkt. 4.8). Periodisk hudundersøkelse anbefales, spesielt for pasienter med risikofaktorer for hudkreft.
Kongestiv hjertesvikt (CHF)
Det har vært rapporter om forverring av kongestiv hjertesvikt (CHF) og nye tilfeller av CHF med TNF -antagonister, inkludert Simponi. Noen tilfeller har dødelig utgang. I en klinisk studie med en annen TNF -antagonist ble det observert forverring av kongestiv hjertesvikt og økt dødelighet på grunn av CHF. Simponi har ikke blitt undersøkt hos pasienter med CHF. Simponi bør brukes med forsiktighet hos pasienter med insuffisiens. Hjertesvikt (NYHA klasse I / II) Pasientene bør overvåkes nøye og Simponi -behandlingen avsluttes hos pasienter som får nye eller forverrede symptomer på hjertesvikt (se pkt. 4.3).
Virkninger på nervesystemet
Bruk av TNF-blokkerende legemidler, inkludert Simponi, har vært assosiert med nye debut eller forverrede tilfeller av kliniske symptomer og / eller radiografiske tegn på demyeliniserende lidelser i sentralnervesystemet, inkludert multippel sklerose og perifere demyeliniseringsforstyrrelser. lidelser, fordeler og risiko ved anti-TNF-behandling bør vurderes nøye før du starter Simponi-behandling.
Avbryt behandling med Simponi bør vurderes hvis disse tilstandene utvikler seg (se pkt. 4.8).
Kirurgiske inngrep
Erfaring med sikkerheten ved Simponi -behandling hos pasienter som har operert, inkludert artroplastikk, er begrenset. Den lange eliminasjonshalveringstiden bør vurderes når du planlegger kirurgi. En pasient som krever operasjon under Simponi -behandling, bør overvåkes nøye for økt risiko for infeksjoner, og passende tiltak bør vurderes.
Immunsuppresjon
Muligheten eksisterer for at anti-TNF-legemidler, inkludert Simponi, påvirker vertens forsvar mot infeksjoner og maligniteter, ettersom TNF formidler betennelse og modulerer cellulær immunrespons.
Autoimmune reaksjoner
Den relative TNF -mangelen? forårsaket av anti-TNF-terapi, kan føre til oppstart av en autoimmun prosess. Hvis en pasient viser symptomer som er prediktive for et lupuslignende syndrom etter behandling med Simponi og er positivt for dobbeltstrengede DNA-antistoffer, må behandlingen med Simponi avbrytes ( se avsnitt 4.8).
Hematologiske reaksjoner
Etter markedsføring har det blitt rapportert tilfeller av pancytopeni, leukopeni, nøytropeni, aplastisk anemi og trombocytopeni hos pasienter behandlet med anti-TNF-legemidler. Cytopenier, inkludert pancytopeni, har ikke blitt rapportert ofte i kliniske studier med Simponi. Alle pasienter bør rådes til å oppsøke lege umiddelbart hvis de utvikler kompatible tegn eller symptomer på bloddyskrasier (f.eks. Vedvarende feber, blåmerker, blødninger og blekhet). Avbryt behandling med Simponi bør vurderes hos pasienter med bekreftet signifikante hematologiske abnormiteter.
Samtidig administrering av TNF -antagonister og anakinra
Alvorlige infeksjoner og nøytropeni har forekommet i kombinasjonskliniske studier av anakinra og en annen TNF -hemmer, etanercept, uten ytterligere klinisk fordel. Gitt bivirkningene som ble observert med denne kombinasjonsbehandlingen, kan lignende toksisiteter oppstå ved kombinasjonen av anakinra og andre TNF -hemmere. Kombinasjonen av Simponi og anakinra anbefales ikke.
Samtidig administrering av TNF -antagonister og abatacept
I kliniske studier var kombinert bruk av TNF-antagonister og abatacept assosiert med økt risiko for infeksjoner, inkludert alvorlige infeksjoner, sammenlignet med TNF-antagonister brukt alene, uten økning i klinisk nytte. Simponi og abatacept anbefales ikke.
Samtidig administrering med andre biologiske behandlinger
Det er utilstrekkelig informasjon om samtidig bruk av Simponi med andre biologiske terapier som brukes til å behandle de samme tilstandene som Simponi. Samtidig bruk av Simponi med disse biologiske stoffene anbefales ikke på grunn av muligheten for økt risiko for infeksjon og andre potensielle legemiddelinteraksjoner.
Erstatning mellom biologiske DMARDer
Forsiktighet bør utvises, og pasientene bør fortsette å overvåkes når de bytter fra en biologisk til en annen, da overlappende biologisk aktivitet kan ytterligere øke risikoen for bivirkninger, inkludert infeksjon.
Vaksinasjoner / smittsomme terapeutiske midler
Pasienter behandlet med Simponi kan få samtidig vaksinasjon, unntatt levende vaksiner (se pkt. 4.5 og 4.6). Hos pasienter behandlet med anti-TNF-terapi er begrensede data tilgjengelig om respons på vaksinasjon, med levende vaksiner eller om sekundær smitteoverføring ved administrering av levende vaksiner. Bruk av levende vaksiner kan føre til kliniske infeksjoner, inkludert infeksjoner som spres .
Andre bruksområder av smittsomme terapeutiske midler som levende svekkede bakterier (for eksempel intravesikale instillasjoner med BCG for kreftbehandling) kan resultere i kliniske infeksjoner, inkludert spredte infeksjoner. Det anbefales at terapeutiske smittestoffer ikke gis samtidig med Simponi.
Allergiske reaksjoner
Etter markedsføring er det rapportert om alvorlige systemiske overfølsomhetsreaksjoner (inkludert anafylaktisk reaksjon) etter administrering av Simponi. Noen av disse reaksjonene oppstod etter første administrering av Simponi. Ved anafylaktiske eller andre allergiske reaksjoner, bør Simponi seponeres umiddelbart og passende terapi startet.
Følsomhet for latex
Nålehetten på den ferdigfylte pennen eller den ferdigfylte sprøyten er laget av latex som inneholder tørket naturgummi og kan forårsake allergiske reaksjoner hos latexfølsomme personer.
Spesielle populasjoner
Eldre (≥ 65 år)
I fase III RA, AP, SA og CU -studiene ble det ikke observert generelle forskjeller i bivirkninger, alvorlige bivirkninger (EAGs) og alvorlige infeksjoner hos pasienter i alderen 65 år eller eldre som fikk behandling. Med Simponi, sammenlignet med yngre pasienter. Imidlertid bør det utvises forsiktighet ved behandling av eldre, og spesiell oppmerksomhet bør rettes mot forekomst av infeksjoner Det var ingen pasienter i alderen 45 år eller eldre i den aksiale SpA -studien.
Nedsatt nyre- og leverfunksjon
Det er ikke utført spesifikke studier med Simponi hos pasienter med nedsatt nyre- eller leverfunksjon. Simponi bør brukes med forsiktighet hos personer med nedsatt leverfunksjon (se pkt.4.2).
Pediatrisk populasjon
Vaksinasjoner
Hvis det er mulig, anbefales det at pediatriske pasienter er oppdatert med alle vaksinasjoner i henhold til gjeldende retningslinjer for immunisering før de starter Simponi-behandling.
Hjelpestoffer
Simponi inneholder sorbitol (E420). Pasienter med sjeldne arvelige problemer med fruktoseintoleranse bør ikke ta Simponi.
Potensial for behandlingsfeil
Simponi er registrert i styrker på 50 mg og 100 mg for subkutan administrasjon. Det er viktig at riktig dose brukes til å administrere riktig dose som angitt i dosering (se pkt.4.2). Det må utvises forsiktighet ved å gi riktig dosering for å sikre at pasientene ikke blir underdosert eller overdosert.
04.5 Interaksjoner med andre legemidler og andre former for interaksjon
Ingen interaksjonsstudier er utført.
Samtidig bruk med andre biologiske behandlinger
Kombinasjonen av Simponi med andre biologiske behandlinger som brukes til å behandle de samme tilstandene som Simponi, inkludert anakinra og abatacept, anbefales ikke (se pkt. 4.4).
Levende vaksiner / smittsomme terapeutiske midler
Levende vaksiner bør ikke gis samtidig med Simponi (se pkt. 4.4 og 4.6).
Smittsomme terapeutiske midler bør ikke administreres samtidig med Simponi (se pkt. 4.4).
Metotreksat
Selv om samtidig bruk av metotreksat (MTX) resulterer i økte simponi -konsentrasjoner ved steady state hos pasienter med RA, AP eller AS, tyder dataene ikke på behov for å justere hverken Simponi -dose eller MTX (se pkt. 5.2).
04.6 Graviditet og amming
Kvinner i fertil alder
Kvinner i fertil alder må bruke tilstrekkelig prevensjon for å forhindre graviditet og fortsette bruken i minst 6 måneder etter den siste golimumab -administrasjonen.
Svangerskap
Det er ingen tilstrekkelige data fra bruk av golimumab hos gravide På grunn av hemming av TNF kan administrering av golimumab påvirke normal immunrespons hos nyfødte. Dyrestudier indikerer ikke direkte eller skadelige effekter. Indirekte effekter på graviditet , embryonisk / fosterutvikling, fødsel eller postnatal utvikling (se pkt. 5.3) Bruk av golimumab anbefales ikke til gravide; golimumab skal bare gis til gravide når det er klart nødvendig.
Golimumab krysser morkaken. Etter behandling med et TNF-hemmende monoklonalt antistoff under graviditet, ble antistoffet funnet i opptil 6 måneder i serumet til spedbarn født av behandlede kvinner.Følgelig kan disse spedbarn ha økt risiko for infeksjon.
Administrering av levende vaksiner hos eksponerte spedbarn i livmoren en golimumab anbefales ikke i 6 måneder etter mors siste golimumab -injeksjon under graviditet (se pkt. 4.4 og 4.5).
Amming
Det er ikke kjent om golimumab skilles ut i morsmelk eller absorberes systemisk etter svelging. Golimumab har vist seg å passere inn i melk hos aper, og fordi humane immunglobuliner skilles ut i melk, bør kvinner ikke amme under behandling og i minst 6 måneder etter behandling med golimumab.
Fruktbarhet
Fertilitetsstudier med golimumab har ikke blitt utført på dyr. En fertilitetsstudie på mus som brukte et lignende antistoff som selektivt hemmer funksjonell aktivitet av murint TNF viste ingen relevante effekter på fruktbarhet (se pkt. 5.3).
04.7 Påvirkning av evnen til å kjøre bil og bruke maskiner
Simponi kan svekke evnen til å kjøre bil og bruke maskiner litt. Svimmelhet kan oppstå etter administrering av Simponi (se pkt. 4.8).
04.8 Bivirkninger
Oppsummering av sikkerhetsprofilen
I kontrollperioden for de sentrale RA-, AP-, SA-, Axial SpA nr- og CU-studiene var infeksjon i øvre luftveier den vanligste bivirkningsreaksjonen (ADR) rapportert hos 12,6% av golimumabbehandlede pasienter. Sammenlignet med 11,0% av kontrollere pasienter. De mest alvorlige bivirkningene som er rapportert for golimumab inkluderer alvorlige infeksjoner (inkludert sepsis, lungebetennelse, TB, invasive soppinfeksjoner og opportunistiske infeksjoner), demyeliniserende sykdommer, HBV-reaktivering, CHF, autoimmune prosesser (lupuslignende syndrom), hematologiske reaksjoner, alvorlig overfølsomhet systemisk ( inkludert anafylaktisk reaksjon), vaskulitt, lymfom og leukemi (se pkt. 4.4).
Tabell med liste over bivirkninger
Bivirkninger observert i kliniske studier og rapportert etter verdensomspennende bruk av golimumab etter markedsføring er oppført i tabell 1. I systemorganklassen er bivirkninger listet etter frekvens ved bruk av følgende kategorier: svært vanlige (≥ 1/10); vanlig (≥ 1/100,
Tabell 1
Tabell med liste over bivirkninger
*: Observert med andre TNF -blokkeringsmidler.
I denne delen presenteres gjennomsnittlig oppfølgingstid (ca. 4 år) for alle golimumab-bruksområder. Der golimumab-bruk er beskrevet etter dose, varierer medianvarigheten av oppfølgingen (ca. 2 år for en 50 mg dose, ca. 3 år for en 100 mg dose) ettersom pasienter kan byttes mellom doser.
Beskrivelse av utvalgte bivirkninger
Infeksjoner
I kontrollperioden for de sentrale studiene var infeksjon i øvre luftveier den vanligste bivirkningen rapportert hos 12,6% av golimumabbehandlede pasienter (forekomst per 100 fagår: 60,8; 95% KI: 55,0, 67,1) sammenlignet med 11,0% av kontrollpasienter (forekomst per 100 personer / år: 54,5; 95% KI: 46,1, 64,0). I de kontrollerte og ukontrollerte fasene av studiene med en median oppfølging på ca. 4 år, var forekomsten per 100 fagår av øvre luftveisinfeksjoner 34,9 hendelser; 95% KI: 33,8, 36,0 for golimumabbehandlede pasienter.
I kontrollperioden for de sentrale studiene ble det observert infeksjoner hos 23,0% av golimumabbehandlede pasienter (forekomst per 100 fagår: 132,0; 95% KI: 123,3, 141,1) sammenlignet med 20, 2% av kontrollpasientene (forekomst pr. 100 fagår: 122,3; 95% KI: 109,5, 136,2). I de kontrollerte og ukontrollerte fasene av studiene med en median oppfølging på ca. 4 år, var forekomsten per 100 fagår av infeksjoner 81,1 hendelser; 95% KI: 79,5, 82,8 for golimumabbehandlede pasienter.
I kontrollperioden for RA-, AP-, SA- og Axial SpA nr-studiene ble det observert alvorlige infeksjoner hos 1,2% av golimumabbehandlede pasienter og 1,2% av kontrollpasientene. Forekomsten av alvorlige infeksjoner per 100 fagår under oppfølgingen i kontrollperioden for RA-, AP-, SA- og nr-Axial SpA-studiene var 7,3; 95% KI: 4,6, 11, 1 for golimumab 100 mg-gruppen , på 2,9; 95% KI: 1,2, 6,0 for golimumab 50 mg -gruppen og 3,6; 95% KI: 1, 5, 7,0 for placebogruppen. I kontrollperioden for golimumab -induksjons -UC -studiene ble det sett alvorlige infeksjoner i 0,8% av golimumabbehandlede pasienter mot 1,5% av kontrollpasientene. Alvorlige infeksjoner som ble sett hos golimumab-behandlede pasienter inkluderte tuberkulose, bakterielle infeksjoner inkludert sepsis og lungebetennelse, invasive soppinfeksjoner og andre opportunistiske infeksjoner. Noen av disse infeksjonene har vært dødelige. I de kontrollerte og ukontrollerte delene av de sentrale studiene med en median oppfølging på opptil 3 år, var det en høyere forekomst av alvorlige infeksjoner, inkludert opportunistiske infeksjoner og TB hos pasienter behandlet med golimumab 100 mg sammenlignet med behandlede pasienter. 50 mg. Forekomsten per 100 fagår for alle alvorlige infeksjoner var 4,1; 95% KI: 3,6, 4,5, for pasienter behandlet med golimumab 100 mg og 2,5; 95% KI: 2,0, 3,1, for pasienter behandlet med golimumab 50 mg.
Ondartede neoplasmer
Lymfom
Forekomsten av lymfom hos golimumabbehandlede pasienter under de sentrale studiene var høyere enn forventet i befolkningen generelt.I de kontrollerte og ukontrollerte delene av disse studiene med en median oppfølging på opptil 3 år, ble det observert en høyere forekomst av lymfom hos pasienter behandlet med golimumab 100 mg sammenlignet med pasienter behandlet med golimumab 50 mg. Lymfom ble diagnostisert hos 11 personer (1 i golimumab 50 mg behandlingsgrupper og 10 i golimumab 100 mg behandlingsgrupper) med forekomst (95% KI) pr. 100 fagår med oppfølging av 0,03 og 0,13 hendelser for henholdsvis golimumab 50 mg og golimumab 100 mg og 0,00 hendelser for placebo. De fleste lymfom forekom i GO-AFTER-studien der pasienter som tidligere var utsatt for anti-TNF-legemidler og med lengre og mer ildfast sykdomsvarighet, ble registrert (se pkt. 4.4).
Andre ondartede neoplasmer enn lymfom
I kontrollperiodene i de sentrale studiene og i omtrent 4 års oppfølging var forekomsten av andre maligniteter enn lymfom (unntatt ikke-melanom hudkreft) lik mellom golimumab og kontrollgrupper. Ca. 4 års oppfølging, forekomsten av ikke-lymfom-maligniteter (unntatt ikke-melanom hudkreft) var lik den i befolkningen generelt.
I de kontrollerte og ukontrollerte periodene av de sentrale studiene med en median oppfølging på opptil 3 år, ble ikke-melanom hudkreft diagnostisert hos 5 personer behandlet med placebo, 10 behandlet med golimumab 50 mg og 31 behandlet med golimumab 100 mg med en forekomst (95% KI) per 100 fagår med oppfølging på 0,36 for kombinert golimumab og 0,87 for placebo.
I de kontrollerte og ukontrollerte periodene av de sentrale studiene med en median oppfølging på opptil 3 år, ble maligniteter i tillegg til melanom, ikke-melanom hudkreft og lymfom diagnostisert hos 5 personer behandlet med placebo, hos 21 behandlet med golimumab 50 mg og hos 34 behandlet med golimumab 100 mg med en forekomst (95% KI) per 100 fagår med oppfølging på 0,48 for kombinert golimumab og 0,87 for placebo (se pkt. 4.4).
Tilfeller rapportert i kliniske studier med nærvær av astma
I en eksplorativ klinisk studie mottok pasienter med alvorlig vedvarende astma en ladningsdose golimumab (150% av den tildelte behandlingsdosen) subkutant i uke 0, etterfulgt av golimumab 200 mg, golimumab 100 mg eller golimumab 50 mg hver. 4 uker subkutant gjennom uke 52. Åtte maligniteter i golimumab kombinasjonsbehandlingsgruppen (n = 230) og ingen i placebobehandlingsgruppen (n = 79) ble rapportert. Lymfom ble rapportert hos 1 pasient, ikke-melanom hudkreft hos 2 pasienter og andre maligniteter hos 5 pasienter. Det var ingen spesifikk sammenslåing av noen form for malignitet.
I den placebokontrollerte fasen av studien var forekomsten (95% KI) av alle maligne sykdommer per 100 fagår av oppfølging 3,19 i golimumabbehandlingsgruppen. Forekomst (95% KI) per 100 fagår av oppfølging -opp hos golimumab-behandlede pasienter var 0,40 for lymfom, 0,79 for ikke-melanom hudkreft og 1,99 for andre maligniteter. For pasienter behandlet med placebo var forekomsten (95% KI) av disse malignitetene per 100 forsøkspersoner / oppfølgingsår 0,00. Betydningen av disse funnene er ukjent.
Nevrologiske hendelser
I de kontrollerte og ukontrollerte periodene av de sentrale studiene med en median oppfølging på opptil 3 år, ble det observert en høyere forekomst av demyelinisering hos pasienter behandlet med golimumab 100 mg sammenlignet med pasienter behandlet med golimumab 50 mg (se pkt. 4.4). .
Økning i leverenzymer
I kontrollperiodene av de sentrale RA- og AP -studiene, små økninger i ALAT (> 1 og 1 og
I kontrollperioden for de sentrale RA- og AS-studiene var ALAT-forhøyelser ≥ 5 ganger ULN uvanlige og ble observert hos et større antall golimumabbehandlede pasienter (0,4%til 0, 9%) sammenlignet med kontrollpasienter (0,0%). Denne trenden ble ikke observert i AP -populasjonen. I de kontrollerte og ukontrollerte periodene av de sentrale RA-, AP- og SA-studiene med en median oppfølging på 5 år, var forekomsten av ALAT-forhøyelser ≥ 5 ganger ULN lik for både golimumabbehandlede og kontrollpasienter. Generelt var disse økningene asymptomatiske, og unormalitetene ble redusert eller løst med fortsettelse eller seponering av golimumab eller modifisering av samtidige medisiner. Ingen tilfeller ble rapportert i de kontrollerte og ukontrollerte periodene i Axial SpA -studien. (Opptil 1 år) I kontrollen perioder av de sentrale CU-induksjonsstudiene med golimumab, ble forhøyelser i ALAT ≥ 5 x ULN observert i lignende frekvenser hos golimumabbehandlede og placebobehandlede pasienter (0, 3% til 1,0%) I de kontrollerte og ukontrollerte periodene av den sentrale UC studier med en median oppfølging på ca. 2 år, var andelen pasienter med ALAT-forhøyelser ≥ 5 x ULN 0,8 % hos pasienter som fikk golimumab i vedlikeholdsperioden av UC-studien.
I de sentrale RA-, AP-, SA- og aksiale SpA-studiene utviklet en pasient i en RA-studie med eksisterende leverabnormaliteter og konfunderende faktormedisiner behandlet med golimumab dødelig ikke-smittsom hepatitt med gulsott. Golimumabs rolle som en medvirkende eller forverrende faktor kan ikke utelukkes.
Reaksjoner på injeksjonsstedet
I kontrollperiodene av de sentrale studiene ble reaksjoner på injeksjonsstedet observert hos 5,4% av golimumabbehandlede pasienter, sammenlignet med 2,0% av kontrollpasientene. Tilstedeværelsen av antistoffer mot golimumab kan øke risikoen for reaksjoner på injeksjonsstedet. De fleste reaksjonene på injeksjonsstedet var milde og moderate, og de hyppigste manifestasjonene var erytem på injeksjonsstedet. Reaksjoner på injeksjonsstedet krever vanligvis ikke seponering av behandlingen med medisinen.
I de kontrollerte fase IIb- og / eller III-studiene med RA, AP, SA, aksial SpA nr, alvorlig vedvarende astma og i fase II / III UC-studier utviklet ingen golimumabbehandlede pasienter anafylaktiske reaksjoner.
Autoimmune antistoffer
I de kontrollerte og ukontrollerte periodene av de sentrale studiene med 1 års oppfølging hadde 3,5% av golimumabbehandlede pasienter og 2,3% av kontrollpasientene nylig positiv ANA (titreringer på 1: 160 eller høyere). Hyppigheten av anti-dsDNA-antistoffer ved 1 års oppfølging hos anti-dsDNA-negative pasienter ved baseline var 1,1%.
Pediatrisk populasjon
Polyartikulær juvenil idiopatisk artritt
Sikkerheten til golimumab ble studert i en fase III -studie av 173 pasienter med pJIA fra 2 til 17 år. Gjennomsnittlig oppfølging var omtrent to år. I denne studien var typen og hyppigheten av rapporterte bivirkninger generelt lik de som ble sett i studier hos voksne med RA.
Rapportering av mistenkte bivirkninger
Rapportering av mistenkte bivirkninger som oppstår etter godkjenning av legemidlet er viktig, ettersom det muliggjør kontinuerlig overvåking av nytte / risiko -balansen for medisinen.Helsepersonell blir bedt om å melde fra om eventuelle mistenkte bivirkninger via Det italienske legemiddelkontoret, nettsted: http://www.agenziafarmaco.gov.it/it/responsabili.
04.9 Overdosering
Enkeltdoser på opptil 10 mg / kg intravenøst ble administrert i en klinisk studie uten dosebegrensende toksisitet. Ved overdosering anbefales det at pasienten overvåkes for tegn og symptomer på bivirkninger og passende symptomatisk behandling iverksettes umiddelbart.
05.0 FARMAKOLOGISKE EGENSKAPER
05.1 Farmakodynamiske egenskaper
Farmakoterapeutisk gruppe: immunsuppressiva, tumornekrosefaktor alfa (TNF-?) -Hemmere, ATC-kode: L04AB06
Virkningsmekanismen
Golimumab er et humant monoklonalt antistoff som danner stabile komplekser med høy affinitet for både de løselige og bioaktive transmembranformene av TNF-? menneskelig, forhindrer binding av TNF-? til reseptorene.
Farmakodynamiske effekter
Golimumab-binding til humant TNF har vist seg å hemme TNF- a Indusert celleoverflateekspresjon av adhesjonsmolekyler, selektin E, vaskulær celleadhesjonsmolekyl type 1 (VCAM) og intracellulært adhesjonsmolekyl av type 1 (ICAM) av humane endotelceller. In vitro, TNF-indusert sekresjon av interleukin (IL) -6, IL-8 og granulocytt- og makrofagkolonistimulerende faktor (GM-CSF) av humane endotelceller ble også hemmet av golimumab.
Det ble observert en forbedring i C-reaktivt protein (CRP) sammenlignet med placebogruppene, og Simponi-behandling resulterte i signifikante reduksjoner i serumnivåer fra baseline av IL-6, ICAM-1, matrix metalloproteinase. 3 (MMP) og vaskulær endotel vekstfaktor (VEGF), sammenlignet med kontrollbehandlingen. Også hos RA- og AS-pasienter er nivåene av TNF-? redusert og IL-8-nivåer redusert hos pasienter med AP. Disse endringene ble observert i den første evalueringen (uke 4) etter den første Simponi -administrasjonen og varte vanligvis gjennom uke 24.
Klinisk effekt
Leddgikt
Effekten av Simponi har blitt demonstrert i tre multisenter, randomiserte, dobbeltblindede, placebokontrollerte kliniske studier utført på mer enn 1500 pasienter ≥ 18 år med moderat til alvorlig aktiv RA diagnostisert i henhold til kriteriene American College of Rheumatology (ACR) ) i minst en 3-måneders periode før screening. Pasientene måtte ha minst 4 hovne og 4 smertefulle ledd. Simponi eller placebo ble administrert subkutant hver 4. uke.
GO-FORWARD evaluerte 444 pasienter med aktiv RA til tross for en stabil dose på minst 15 mg MTX per uke og som ikke tidligere hadde blitt behandlet med anti-TNF-medisiner. Pasientene ble randomisert til placebo + MTX, Simponi 50 mg + MTX, Simponi 100 mg + MTX eller Simponi 100 mg + placebo. Pasienter som fikk placebo + MTX ble etter uke 24 tildelt Simponi 50 mg + MTX. I uke 52 gikk pasientene inn i en åpen langsiktig forlengelsesstudie.
GO-AFTER evaluerte 445 pasienter som tidligere ble behandlet med ett eller flere anti-TNF-legemidler, adalimumab, etanercept eller infliximab. Pasientene ble randomisert til placebo, Simponi 50 mg eller Simponi 100 mg. Under studien kunne pasientene fortsette samtidig DMARD -behandling med MTX, sulfasalazin (SSZ) og / eller hydroksyklorokin (HCQ). Årsakene til seponering av tidligere anti-TNF-behandlinger var mangel på effekt (58%), intoleranse (13%) og / eller andre årsaker enn sikkerhet eller effekt (29%, hovedsakelig av økonomiske årsaker).
GO-BEFORE evaluerte 637 pasienter med aktiv RA, MTX-naive og ikke tidligere behandlet med et anti-TNF-legemiddel. Pasientene ble randomisert til å motta placebo + MTX, Simponi 50 mg + MTX, Simponi 100 mg + MTX eller Simponi 100 mg + placebo. I uke 52 gikk pasientene inn i en åpen langtidsstudie hvor pasienter som fikk placebo + MTX og hadde minst 1 smertefull eller hovent ledd ble flyttet til Simponi 50 mg + MTX-behandling.
I GO-FORWARD var de (co) primære endepunktene prosentandelen av pasientene som oppnådde en ACR 20-respons i uke 14 og forbedring i Health Assessment Questionnaire (HAQ) i uke 24. fra baseline. I GO-EFTER var det primære endepunktet andelen av pasienter som oppnådde en ACR 20-respons i uke 14. I GO-FEFORE var de co-primære endepunktene andelen pasienter som oppnådde en ACR 50-respons i uke 24 og en endring fra baseline i modifisert Sharp-score av van der Heijde (vdH -S) i uke 52. I tillegg til de primære endepunktene ble det utført ytterligere vurderinger av virkningen av Simponi-behandling på tegn og symptomer på leddgikt, radiografisk respons, fysisk funksjon og statusrelatert livskvalitet. Av helse.
Generelt ble det ikke observert noen klinisk signifikante forskjeller i effektvurderinger mellom Simponi 50 mg og 100 mg i kombinasjon med MTX doseringsregimer opp til uke 104 i GO-FORWARD og GO-BEFORE og opptil uke 24. i GO-EFTER I hver av RA-studiene i henhold til studiedesignet, kunne pasienter i langtidsforlengelsen byttes mellom Simponi 50 mg og 100 mg doser etter studielege.
Tegn og symptomer
De viktigste ACR-kriteriene for Simponi 50 mg dose ved uke 14, 24 og 52 for GO-FORWARD, GO-AFTER og GO-BEFORE er vist i tabell 2 og er beskrevet nedenfor. Respons ble sett i den første evalueringen (uke 4) etter den første administreringen av Simponi.
I GO-FORWARD-studien, av de 89 individene randomisert til Simponi 50 mg + MTX, var 48 fortsatt i behandling i uke 104. Blant disse hadde 40, 33 og 24 pasienter ACR 20/50/70 respons i uke 104, henholdsvis Blant pasienter som ble værende i studien og behandlet med Simponi, ble lignende ACR 20/50/70 responsfrekvenser observert fra uke 104 til uke 256.
I GO-AFTER-studien var prosentandelen pasienter som oppnådde en ACR 20-respons høyere blant Simponi-behandlede pasienter sammenlignet med placebobehandlede pasienter, uavhengig av den rapporterte årsaken til at en eller flere antiinflammatoriske behandlinger ble avbrutt. -TNF.
Tabell 2
Viktige effektresultater fra de kontrollerte delene av GO-FORWARD, GO-AFTER og GO-BEFORE-studiene
a n tilsvarer randomiserte pasienter; det faktiske antallet pasienter som kan evalueres for hvert endepunkt kan variere etter tidspunkt.
* p ≤ 0,001
NA: Gjelder ikke
I GO-BEFORE-studien var den primære analysen hos pasienter med moderat til alvorlig revmatoid artritt (Simponi 50 og 100 mg + MTX kombinasjonsgrupper versus MTX alene for ACR 50) ikke statistisk signifikant i uke 24 (p = 0,053 I uke 52 på tvers befolkningen, var andelen pasienter i Simponi 50 mg + MTX -gruppen som oppnådde en ACR -respons generelt høyere, men ikke signifikant forskjellig sammenlignet med MTX alene (se tabell 2.) Ytterligere undergruppeanalyser representativ for den indikerte populasjonen av pasienter med alvorlig , aktiv og progressiv RA.En generelt overlegen effekt ble påvist med Simponi 50 mg + MTX versus MTX alene i den angitte populasjonen sammenlignet med den totale populasjonen.
I GO-FORWARD og GO-AFTER-studiene ble det observert statistisk og klinisk signifikante responser på sykdomsaktivitetsskalaen (DAS28) på hvert forhåndsspesifisert stadium, i uke 14 og uke 24 (s ≤ 0,001). Blant pasientene som ble værende på Simponi -behandling, randomisert ved studiestart, ble DAS28 -responser opprettholdt gjennom uke 104. Blant pasienter som ble værende i studien og behandlet med Simponi, var DAS28 -svar like fra uke 104 til uke 256.
I GO-BEFORE-studien ble større klinisk respons vurdert, definert som å opprettholde en ACR 70-respons over en kontinuerlig periode på 6 måneder. I uke 52 oppnådde 15% av pasientene i Simponi 50 mg + MTX -gruppen en overlegen klinisk respons sammenlignet med 7% av pasientene i placebo + MTX -gruppen (p = 0,018). Av de 159 pasientene som ble randomisert til Simponi 50 mg + MTX, var 96 fortsatt i behandling i uke 104. Blant disse hadde 85, 66 og 53 pasienter henholdsvis ACR 20/50/70 respons i uke 104, og behandlet med Simponi, tilsvarende ACR 20/50/70 responsrater ble observert fra uke 104 til uke 256.
Radiografisk respons:
I GO-BEFORE-studien ble endringer fra grunnlinjen i vdH-S-score, en sammensatt strukturell skadescore som radiografisk måler antall og størrelse på ledderosjoner og graden av fellesromreduksjon i hender / håndledd og føtter, brukt til å vurdere graden av strukturell skade. Nøkkelresultater for Simponi ved 50 mg i uke 52 er presentert i tabell 3.
Antall pasienter uten ny erosjon eller endring fra baseline i total vdH-S-score ≤ 0 var signifikant høyere i Simponi-gruppen enn i kontrollgruppen (p = 0,003). Radiografiske effekter observert i uke 52 ble opprettholdt gjennom uke 104. Blant pasienter som ble værende i studien og behandlet med Simponi, var radiografiske effekter like fra uke 104 til uke 256.
Tabell 3
Gjennomsnitt (SD) for radiografiske endringer fra baseline til uke 52 i total vdH-S-score for GO-BEFORE studiepopulasjonen
a n tilsvarer randomiserte pasienter
* p = 0,015
** p = 0,044
Fysisk funksjon og helserelatert livskvalitet
Fysisk funksjon og funksjonshemming ble vurdert som separate endepunkter i GO-FORWARD og GO-AFTER-studiene, ved bruk av HAQ DI funksjonshemmingindeks. I disse studiene, i uke 24, viste Simponi en klinisk og statistisk signifikant forbedring i HAQ DI fra baseline sammenlignet med kontrollgruppen. Blant pasienter som forble på Simponi -behandling, randomisert ved studiestart, ble forbedring i HAQ DI opprettholdt gjennom uke 104 Blant pasienter som ble værende i studien og behandlet med Simponi, var forbedring i HAQ DI lik fra uke 104 til uke 256.
I GO-FORWARD-studien ble det vist klinisk og statistisk signifikant forbedring av helserelatert livskvalitet målt ved den fysiske komponentpoengsummen til SF-36 hos pasienter behandlet med Simponi versus placebo i uke 24. Blant pasienter som ble værende i Simponi-behandling , randomisert til studiestart, ble forbedringen i SF-36 opprettholdt gjennom uke 104.Blant pasientene som ble værende i studien og behandlet med Simponi, var forbedringen i den fysiske komponenten i SF-36 lik fra uke 104 til og med uke 256. I GO-FORWARD og GO-AFTER-studiene ble det observert statistisk signifikante forbedringer i ". "tretthet, i henhold til FACIT-F (Functional Assessment of Chronic Illness Therapy-Fatigue) skala.
Psoriasisartritt
Effekten og sikkerheten til Simponi ble evaluert i en multisenter, randomisert, dobbeltblind, placebokontrollert klinisk studie (GO-REVEAL) utført på 405 voksne pasienter med aktiv PA (≥ 3 hovne ledd og ≥ 3 leddsmerter), til tross for behandling med ikke-steroide antiinflammatoriske legemidler (NSAIDs) eller DMARDs. Pasienter i denne studien hadde diagnosen AP i minst 6 måneder og minst mild psoriasis. Pasienter med hver subtype psoriasisartritt, inkludert leddgikt, ble registrert polyartikulære uten revmatoid knuter (43%), asymmetrisk perifer leddgikt (30%), distal interphalangeal leddgikt (DIP) (15%), spondylitt med perifer leddgikt (11%) og lemlestende leddgikt (1%). ingen tidligere behandling med anti-TNF Simponi eller placebo ble administrert subkutant hver 4. uke. Pasientene ble randomisert til placebo, Simponi 50 mg eller til S pålegge 100 mg. Pasienter som fikk placebo ble etter uke 24 tildelt Simponi 50 mg. I uke 52 gikk pasientene inn i en langsiktig åpen forlengelsesstudie.
Omtrent 48% av pasientene fortsatte med stabile doser metotreksat (≤ 25 mg / uke). De ko-primære endepunktene var andelen pasienter som oppnådde en ACR 20-respons i uke 14 og endringen fra baseline i endret AP vdH-S total score i uke 24.
Generelt ble det ikke observert noen klinisk signifikante forskjeller i effektmålinger mellom Simponi 50 mg og 100 mg doseringsregimer gjennom uke 104. I følge studiens design kan pasienter i den langsiktige forlengelsen gjennomgå en veksling mellom Simponi 50 mg og 100 mg doser etter studielege.
Tegn og symptomer
Nøkkelresultater for 50 mg dosen i uke 14 og 24 er vist i tabell 4 og er beskrevet nedenfor.
Tabell 4
Viktige effektresultater fra GO-REVEAL-studien
* s
a n tilsvarer randomiserte pasienter; det faktiske antallet pasienter som kan evalueres for hvert endepunkt kan variere etter tidspunkt
b Psoriasisområde og alvorlighetsindeks
c Basert på undergruppen av pasienter med kroppsoverflate (BSA) involvering ≥ 3%ved baseline, 79 pasienter (69,9%) i placebogruppen og 109 (74,3%) i Simponi 50 mg.
Respons ble observert ved den første evalueringen (uke 4) etter den første administreringen av Simponi. Lignende ACR 20 -responser ble funnet i uke 14 hos pasienter med polyartikulær artritt i fravær av reumatoid knuter og AP -subtyper, asymmetrisk perifer leddgikt. Antallet pasienter med andre PA -undertyper var for lite til å gi en meningsfull vurdering. Svarene observert i Simponi -behandlingsgruppene var like hos pasienter som ble behandlet eller ikke med samtidig MTX. Av de 146 pasientene som ble randomisert til Simponi 50 mg, var 70 fortsatt i behandling i uke 104. Blant disse 70 pasientene hadde henholdsvis 64, 46 og 31 pasienter en ACR 20/50/70 respons. Blant pasienter som ble værende i studien og behandlet med Simponi, ble lignende ACR 20/50/70 responsfrekvenser observert fra uke 104 til uke 256.
Statistisk signifikante responser ble også observert i DAS28 i uke 14 og 24 (s
I uke 24 ble det observert forbedringer i perifere aktivitetsparametere som er typiske for psoriasisartritt (f.eks. Antall hovne ledd, antall ømme ledd, daktylitt og entesitt) hos pasienter behandlet med Simponi. Behandling med Simponi resulterte i betydelig forbedring av fysisk funksjon som vurdert av HAQ DI og betydelige forbedringer i helserelatert livskvalitet basert på sammendrag av de fysiske og mentale komponentene i SF-36. På Simponi-behandling, som de ble randomisert til ved studiestart ble DAS28- og HAQ DI -responsene opprettholdt gjennom uke 104. Blant pasientene som ble værende i studien og behandlet med Simponi, var svarene DAS28 og HAQ DI uke 104 til uke 256.
Radiografisk respons:
Strukturell skade på hender og føtter ble radiologisk vurdert av endringen fra baseline i vdH-S-poengsummen, modifisert for AP med tillegg av de distale interphalangeale leddene (DIP) i hånden.
Simponi 50 mg behandling reduserer progresjonshastigheten for perifer leddskade sammenlignet med placebobehandling i uke 24 målt som endring fra baseline i modifisert total vdH -S -score (gjennomsnitt ± SD -score var 0,27 ± 1, 3 i placebogruppen sammenlignet med - 0,16 ± 1,3 i Simponi -gruppen; p = 0,011). Av de 146 pasientene som ble randomisert til Simponi 50 mg, var røntgendata i uke 52 tilgjengelig for 126 pasienter, hvorav 77% ikke viste noen progresjon fra baseline. I uke 104 var røntgendata tilgjengelig for 114 pasienter og 77% viste ingen progresjon fra baseline. Blant pasienter som ble værende i studien og behandlet med Simponi, viste tilsvarende andeler av pasientene ingen progresjon fra baseline fra uke 104 til uke 256.
Aksial spondylartritt
Ankyloserende spondylitt
Effekten og sikkerheten til Simponi ble evaluert i en multisenter, dobbeltblind, randomisert, placebokontrollert klinisk studie (GO-RAISE) utført på 356 voksne pasienter med aktiv ankyloserende spondylitt (definert som Bath Index patologisk aktivitet ved ankyloserende spondylitt (BASDAI) ) ≥ 4 og en VAS for totale ryggsmerter ≥ 4 på en skala fra 0 til 10 cm). Pasienter påmeldt denne studien hadde sykdommen i aktiv fase, til tross for nåværende eller tidligere behandling med NSAIDs eller DMARDs og ikke tidligere hadde blitt behandlet med anti-TNF-legemidler. Simponi eller placebo ble administrert subkutant hver 4. uke. Pasientene ble randomisert til placebo, Simponi 50 mg eller Simponi 100 mg og kunne fortsette samtidig DMARD -behandling (MTX, SSZ og / eller HCQ). Det primære endepunktet var prosentandelen av pasientene med ankyloserende spondylittvurderingsstudiegruppens (ASAS 20) respons i uke 14. Placebokontrollerte effektdata ble samlet inn og analysert gjennom uke 24.
Nøkkelresultatene for dosen på 50 mg er vist i tabell 5 og er beskrevet nedenfor. Generelt ble det ikke observert noen klinisk signifikante forskjeller i effektmål mellom Simponi 50 mg og 100 mg doseringsregimer opp til uke 24. I følge studiens utforming kunne pasienter i den langsiktige forlengelsen gjennomgå en bytte mellom Simponi 50 mg og 100 mg doser etter studielege.
Tabell 5
Viktige effektresultater fra GO-RAISE-studien
* p ≤ 0,001 for alle sammenligninger
a n tilsvarer randomiserte pasienter; det faktiske antallet pasienter som kan evalueres for hvert endepunkt kan variere etter tidspunkt
Blant pasientene som ble værende i studien og behandlet med Simponi, var andelen pasienter med ASAS 20 og ASAS 40 respons lik fra uke 24 til uke 256.
Statistisk signifikante responser ble også observert i BASDAI 50, 70 og 90 (p ≤ 0,017) i uke 14 og 24. Forbedringer i viktige sykdomsaktivitetsmålinger ble funnet ved den første vurderingen (uke 4) etter den første Simponi -administrasjonen som ble opprettholdt gjennom uken 24. Blant pasienter som ble værende i studien og behandlet med Simponi, ble lignende endringshastigheter observert fra BASDAI fra uke 24 til uke 256. "Konsekvent effekt ble observert hos pasienter uavhengig av bruk av DMARD (MTX, sulfasalazin og / eller hydroksyklorokin), tilstedeværelse av HLA-B27-antigenet eller CRP-nivåer ved baseline basert på vurderingen av ASAS 20-responser i uke 14.
Simponi-behandling resulterte i betydelige forbedringer i fysisk funksjon, målt ved endringer fra baseline i Bath Ankylosing Spondylitis Functional Index (BASFI) i uke 14 og 24. Helserelatert livskvalitet, målt ved komponentpoeng SF-36, var signifikant forbedret i uke 14 og 24. Blant pasientene som ble værende i studien og behandlet med Simponi, var forbedringer i fysisk funksjon og helserelatert livskvalitet like fra uke 24 til uke 256.
Ikke-radiografisk aksial spondylartritt
Sikkerhet og effekt av Simponi ble evaluert i en multisenter, randomisert, dobbeltblind, placebokontrollert studie (GO-AHEAD) utført på 197 voksne pasienter med alvorlig aksial SpA nr i aktiv fase (definert som de pasientene som oppfylte ASAS-klassifiseringen kriterier for diagnose av aksial spondylartritt, men oppfylte ikke de modifiserte New York -kriteriene for diagnosen AS.) Pasienter som var påmeldt denne studien hadde aktiv sykdom (definert av en BASDAI ≥ 4 og en Visual Analogue Scale (VAS) for generelle ryggsmerter) ≥ 4, hver på en skala fra 0 til 10 cm) til tross for pågående eller tidligere NSAID -behandling og ikke tidligere hadde blitt behandlet med noe biologisk middel inkludert anti -TNF. Pasientene ble randomisert til placebo eller Simponi 50 mg administrert subkutant hver 4. uke. Kl. uke 16 kom pasienter inn en åpen behandlingsperiode der alle fikk Simponi 50 mg administrert subkutant hver 4. uke gjennom uke 48 med effektvurderinger utført gjennom uke 52 og sikkerhetsoppfølging gjennom uke 60. Omtrent 93% av pasientene som hadde fått Simponi i begynnelsen av den åpne forlengelsen (uke 16) forble i behandling til slutten av studien (uke 52). Analyser ble utført både på hele populasjonen som ble behandlet (AT, N = 197) og på befolkningen med objektive tegn på betennelse (OSI, N = 158, som indikert av høye nivåer av CRP og / eller ved "tegn på sacroiliitt i MR utført ved baseline) Placebokontrollerte effektdata ble samlet inn og analysert gjennom uke 16.Det primære endepunktet var andelen pasienter som oppnådde en ASAS 20 -respons i uke 16. Viktige resultater er vist i tabell 6 og er beskrevet nedenfor.
Tabell 6
Viktige effektresultater fra GO-AHEAD-studien i uke 16
a n tilsvarer randomiserte og behandlede pasienter
b Ankyloserende spondylitt C-reaktiv proteinsykdomsaktivitetspoeng (AT-Placebo, N = 90; AT-Simponi 50 mg, N = 88; OSI-Placebo, N = 71; OSI-Simponi 50 mg, N = 71)
c n tilsvarer antall pasienter med baseline og uke 16 MR -data
d SPARCC (Spondyloarthritis Research Consortium of Canada)
** p vs placebo
* p vs placebo
Statistisk signifikante forbedringer i tegn og symptomer på alvorlig aktiv fase nr aksial SpA ble påvist hos pasienter behandlet med Simponi 50 mg sammenlignet med placebo i uke 16 (tabell 6). Forbedringer ble sett ved den første evalueringen (uke 4) etter den første administreringen av Simponi. SPARCC MR -poengsummen viste statistisk signifikante reduksjoner i betennelse i SI -leddet i uke 16 hos pasienter behandlet med Simponi 50 mg sammenlignet med placebo (tabell 6). Smerter vurdert av VAS generelle ryggsmerter og nattlige ryggsmerter og sykdomsaktivitet målt ved ASDAS-C viste også statistisk signifikant forbedring fra baseline til uke 16 hos pasienter behandlet med Simponi 50 mg sammenlignet med placebo (p
Statistisk signifikante forbedringer i spinal mobilitet som vurdert av BASMI (Bath Ankylosing Spondylitis Metrology Index) og i fysisk funksjon som vurdert av BASFI ble påvist hos pasienter behandlet med Simponi 50 mg sammenlignet med pasienter behandlet med placebo (p
For alle endepunktene beskrevet ovenfor ble det også påvist statistisk signifikante resultater i OSI -populasjonen i uke 16.
Både i AT- og OSI -populasjonen vedvarte forbedringene i tegn og symptomer, ryggradsmobilitet, fysisk funksjon, livskvalitet og produktivitet observert i uke 16 blant pasienter behandlet med Simponi 50 mg hos de pasientene som var igjen i studien. I uke 52.
Ulcerøs kolitt
Effekten av Simponi ble evaluert i to randomiserte, dobbeltblindede, placebokontrollerte kliniske studier på voksne pasienter.
Induksjonsstudien (PURSUIT-induksjon) evaluerte pasienter med moderat til alvorlig aktiv ulcerøs kolitt (Mayo score 6 til 12; endoskopisk sub-score ≥ 2) som hadde utilstrekkelig respons eller som ikke hadde tolerert konvensjonelle behandlinger eller at de var kortikosteroidavhengige. I dosebekreftelsesdelen av studien ble 761 pasienter randomisert til å motta Simponi SC 400 mg i uke 0 og 200 mg i uke 2, Simponi SC 200 mg i uke 0 og 100 mg i uke 2 eller placebo SC i uke 0 og 2. Samtidig administrering av stabile doser av orale aminosalicylater, kortikosteroider og / eller immunmodulerende midler var tillatt. Effekten av Simponi fram til uke 6 ble evaluert i denne studien.
Resultatene av vedlikeholdsstudien (PURSUIT-Maintenance) var basert på evalueringen av 456 pasienter som hadde oppnådd en klinisk respons i forrige Simponi-induksjon. Pasientene ble randomisert til å få Simponi 50 mg, Simponi 100 mg eller placebo administrert subkutant hver 4. uke. Samtidig administrering av stabile doser av orale aminosalicylater og / eller immunmodulerende midler var tillatt. Kortikosteroider skulle gradvis reduseres ved starten av vedlikeholdsstudien. Effekten av Simponi ble evaluert opp til uke 54 i denne studien. Pasienter som hadde fullført vedlikeholdsstudien opp til uke 54 fortsatte behandlingen i en studie. Forlengelse, med effekt vurdert opp til uke 216. Evaluering av effekt i forlengelsesstudien var basert på endringer i bruk av kortikosteroider, legers globale vurdering (PGA) av sykdomsaktivitet og forbedring av livskvalitet målt ved spørreskjemaet Inflammatory Towel Disease (IBDQ).
Tabell 7
Viktige effektresultater fra undersøkelsene PURSUIT - Induction og PURSUIT - Maintenance
N = antall pasienter
** p ≤ 0,001
* p ≤ 0,01
a Defineres som en nedgang fra baseline i Mayo-score ≥ 30% og ≥ 3 poeng, ledsaget av en nedgang i sub-score for endetarmsblødning ≥ 1 eller en sub-score for rektal blødning på 0 eller 1.
b Definert som Mayo-score ≤ 2 poeng, uten individuelle delpoeng> 1
c Definert som 0 eller 1 i den endoskopiske delpoengsummen til Mayo-poengsummen.
d Bare induksjon med Simponi.
e Pasientene ble evaluert for CU -aktivitet med Mayo -partiescore hver 4. uke (tap av respons ble bekreftet med endoskopi). Derfor var en pasient som opprettholdt responsen i en kontinuerlig klinisk responsstilstand ved hver klinisk evaluering gjennom uke 54.
f En pasient måtte være i remisjon i uke 30 og 54 (viser ingen tap av respons på noe tidspunkt opp til uke 54) for å oppnå vedvarende remisjon.
g Hos pasienter som veide mindre enn 80 kg, viste en høyere andel av pasientene som fikk 50 mg vedlikeholdsterapi vedvarende klinisk remisjon enn de som fikk placebo.
Flere Simponi-behandlede pasienter viste vedvarende slimhinneheling (pasienter med slimhinneheling i uke 30 og 54) i 50 mg-gruppen (42%, nominell p
Blant de 54% av pasientene (247/456) som fikk samtidig kortikosteroidbehandling ved starten av PURSUIT-vedlikehold, var andelen pasienter som opprettholdt en klinisk respons gjennom uke 54 og ikke mottok samtidig kortikosteroidbehandling ved starten av PURSUIT-vedlikehold . uke 54 var større i 50 mg -gruppen (38%, 30/78) og i 100 mg -gruppen (30%, 25/82) sammenlignet med placebogruppen (21%, 18/87). Andelen pasienter som hadde eliminert kortikosteroider i uke 54 var større i 50 mg -gruppen (41%, 32/78) og i 100 mg -gruppen (33%, 27/82) sammenlignet med placebogruppen (22%, 19/87). hadde deltatt i forlengelsesstudien, ble andelen personer som forble kortikosteroidfrie generelt opprettholdt gjennom uke 216.
I uke 6 forbedret Simponi livskvaliteten signifikant målt ved endring fra baseline i et sykdomsspesifikt tiltak, IBDQ (Inflammatory Towel Disease Questionnaire). Blant pasienter som fikk vedlikeholdsbehandling med Simponi, var forbedringen av livskvalitet målt ved IBDQ ble opprettholdt gjennom uke 54.
Omtrent 63% av pasientene som fikk Simponi ved starten av forlengelsesstudien (uke 56) forble behandling til slutten av studien (siste administrering av golimumab i uke 212).
Immunogenisitet
I fase III RA-, AP- og SA-studiene gjennom uke 52 ble antistoffer mot golimumab påvist med enzymet immunoassay (EIA) hos 5% (105/2062) av golimumabbehandlede pasienter, og der testet var nesten alle antistoffer som nøytraliserer in vitro. Lignende prosenter har blitt fremhevet i de revmatologiske indikasjonene. Samtidig administrering av MTX resulterte i en lavere prosentandel av pasientene med antistoffer mot golimumab enn pasienter som fikk golimumab uten MTX (henholdsvis ca. 3% [41/1235] mot 8% [64/827]).
I aksial SpA nr ble antistoffer mot golimumab påvist hos 7% (14/193) av golimumabbehandlede pasienter gjennom uke 52 med EIA-analysen.
I fase II og III UC-studier gjennom uke 54 ble antistoffer mot golimumab påvist med EIA-analysen hos 3% (26/946) av golimumabbehandlede pasienter. Sekstiåtte prosent (21/31) av de antistoff-positive pasientene hadde nøytraliserende antistoffer in vitro. Samtidig behandling med immunmodulatorer (azatioprin, 6-merkaptopurin og MTX) resulterte i en lavere andel pasienter med antistoffer mot golimumab enn hos pasienter som fikk golimumab uten immunmodulatorer (henholdsvis 1% (4/308) versus 3% (22). / 638)). Blant pasienter som fortsatte forlengelsesstudien og som hadde evaluerbare prøver fram til uke 228, ble det påvist antistoffer mot golimumab hos 4% (23/604) av golimumabbehandlede pasienter. 82 prosent (18/22) av de antistoff-positive pasientene hadde nøytraliserende antistoffer in vitro.
En legemiddel-tolerant EIA-analyse ble brukt i pIA-studien for påvisning av antistoffer mot golimumab. Gitt den økte sensitiviteten og forbedrede legemiddeltoleransen, forventet det en høyere forekomst av antistoffer mot golimumab med EIA-analysen. Legemiddel-tolerant kontra EIA. I fase III-pIA-studien gjennom uke 48 ble antistoffer mot golimumab påvist med stofftolerant EIA-analyse hos 40% (69/172) av golimumabbehandlede barn, hvorav de fleste hadde en titer mindre enn 1: 1000. En effekt på golimumab -serumkonsentrasjoner ble sett ved titre> 1: 100, mens det ikke ble sett effekt på effekt opp til titre> 1: 1000, selv om barnetall med titre> 1: 1000 var lavt (N = 8). Blant barn som testet positivt for antistoffer mot golimumab, hadde 39% (25/65) nøytraliserende antistoffer. Den høyere forekomsten av antistoffer med den legemiddel-tolerante EIA-analysen, med tanke på at de hovedsakelig var antiterer med lav titer, hadde ingen åpenbar innvirkning på legemiddelnivå, effekt og sikkerhet og representerer derfor ingen nye sikkerhetssignaler.
Tilstedeværelsen av antistoffer mot golimumab kan øke risikoen for reaksjoner på injeksjonsstedet (se pkt. 4.4). Det lille antallet positive pasienter med golimumab-antistoff begrenser muligheten til å trekke faste konklusjoner om forholdet mellom antigolimumab-antistoffer og klinisk effekt eller sikkerhetstiltak.
Siden immunogenisitetsanalyser er produkt- og analysespesifikke, er sammenligning av antistoffprosent med andre produkter ikke hensiktsmessig.
Pediatrisk populasjon
Polyartikulær juvenil idiopatisk artritt
Sikkerhet og effekt av Simponi ble evaluert i en randomisert, dobbeltblind, placebokontrollert suspensjonsstudie (GO-KIDS) hos 173 barn (2 til 17 år) med aktiv pJIA med minst 5 aktive ledd. Og utilstrekkelig respons på MTX. Barn med polyartikulær kur JIA (revmatoid faktor positiv eller negativ polyartritt, omfattende oligoartritt, ung psoriasisartritt eller systemisk JIA uten pågående systemiske symptomer) ble inkludert i studien. Median antall aktive ledd ved baseline var 12 og median CRP var 0,17 mg / dL.
Del 1 av studien involverte en åpen 16-ukers fase der de 173 påmeldte barna fikk Simponi 30 mg / m2 (maks 50 mg) subkutant hver 4. uke og MTX. De 154 barna som oppnådde en ACR Ped 30 -respons i uke 16 gikk inn i del 2 av studien, den randomiserte abstinensfasen, og fikk Simponi 30 mg / m2 (maks 50 mg) + MTX eller placebo + MTX hver. 4 uker. Etter sykdomsbluss fikk barna Simponi 30 mg / m2 (maks. 50 mg) + MTX. I uke 48 gikk babyene inn i en langsiktig forlengelsesfase.
Barna i denne studien viste ACR Ped 30, 50, 70 og 90 svar allerede i uke 4.
I uke 16 var 87% av barna henholdsvis ACR Ped 30 -respondere og 79%, 66% og 36% av barna var henholdsvis ACR Ped 50, ACR Ped 70 og ACR Ped 90. I uke 16 var 34 % av barna hadde inaktiv sykdom definert av tilstedeværelsen av alle følgende: fravær av ledd med aktiv leddgikt; fravær av feber, utslett, serositis, splenomegali, hepatomegali eller generalisert lymfadenopati som kan tilskrives JIA, fravær av aktiv uveitt; Normal ESR (
I uke 16 viste alle ACR Ped -komponenter klinisk relevant forbedring fra baseline (se tabell 8).
Tabell 8
Forbedringer fra baseline i ACR Ped -komponenter i uke 16
ved baseline = uke 0
b "n" gjenspeiler registrerte pasienter
c VAS: Visual Analog Scale
d CHAQ: Spørreskjema for vurdering av barns helse
og ESR (mm / t): sedimentasjonshastighet for erytrocytter (millimeter per time)
Studiens primære endepunkt, prosentandelen barn som var ACR Ped 30 -respondenter i uke 16 og som ikke opplevde en forverring mellom uke 16 og uke 48, ble ikke oppfylt.De fleste av barna hadde ikke opplevd en forverring.forverring mellom uke 16 og uke 48 (59% i Simponi + MTX -gruppen og 53% i henholdsvis placebo + MTX -gruppen; p = 0,41).
En forhåndsspesifisert undergruppeanalyse av det primære endepunktet basert på baseline CRP-verdier (≥ 1 mg / dL vs.
I uke 48 var 53% og 55% av barna i Simponi + MTX -gruppen og placebo + MTX -gruppen henholdsvis ACR Ped 30 -respondere og 40% og 28% av barna i Simponi + MTX -gruppen og placebo + MTX oppnådde henholdsvis inaktiv sykdom.
Det europeiske legemiddelkontoret (European Medicines Agency) har utsatt forpliktelsen til å levere resultatene av studier med Simponi hos den pediatriske populasjonen ved ulcerøs kolitt (se pkt. 4.2 for informasjon om pediatrisk bruk).
05.2 Farmakokinetiske egenskaper
Absorpsjon
Etter en enkelt "subkutan golimumab -administrering til friske personer eller RA -pasienter, varierte gjennomsnittlig tid for å nå maksimale serumkonsentrasjoner (Tmax) fra 2 til 6 dager. En" 50 mg subkutan golimumab -injeksjon hos friske personer resulterte i en gjennomsnittlig maksimal serumkonsentrasjon (Cmax) ± standardavvik på 3,1 ± 1,4 mcg / ml.
Etter en enkelt 100 mg subkutan injeksjon var golimumabs absorpsjon lik i arm, mage og lår, med en gjennomsnittlig absolutt biotilgjengelighet på 51%. Subkutant forventes den absolutte biotilgjengeligheten av en 50 mg eller 200 mg golimumab dose å være lik.
Fordeling
Etter en enkelt IV -administrering var gjennomsnittlig distribusjonsvolum 115 ± 19 ml / kg.
Eliminering
Systemisk clearance av golimumab ble estimert til 6,9 ± 2,0 ml / dag / kg. Den terminale halveringstiden ble estimert til å være ca. 12 ± 3 dager hos friske personer og hadde lignende verdier hos pasienter med RA, AP, SA eller CU.
Når en 50 mg dose golimumab ble administrert subkutant til pasienter med RA, AP eller AS hver fjerde uke, nådde serumkonsentrasjonene steady state etter uke 12. Ved samtidig bruk av MTX ga behandling med golimumab 50 mg subkutant hver 4. uke et gjennomsnitt (± standardavvik) gjennom serumkonsentrasjon ved steady state på ca. 0,6 ± 0,4 μg / ml hos pasienter med aktiv RA til tross for MTX -behandling, ca. 0,5 ± 0,4 μg / ml hos pasienter med aktiv PA og ca. 0,8 ± 0,4 μg / ml hos AS -pasienter Golimumab gjennomsnittlig steady state gjennom serumkonsentrasjoner hos pasienter med Axial SpA nr var lik de som ble observert hos AS -pasienter etter subkutan administrering av golimumab 50 mg hver 4. uke.
Pasienter med RA, AP eller SA som ikke fikk MTX samtidig administrert, hadde omtrent 30% lavere steady state golimumab-konsentrasjoner enn hos pasienter som fikk golimumab med MTX. Hos et begrenset antall RA-pasienter behandlet med golimumab subkutant i mer enn 6 måneder, reduserte samtidig administrering av MTX den tilsynelatende clearance av golimumab med omtrent 36%. Imidlertid indikerer populasjonsfarmakokinetiske analyser at samtidig bruk av NSAID, orale kortikosteroider eller sulfasalazin ikke påvirket den tilsynelatende clearance av golimumab.
Etter induksjonsdoser på 200 mg og 100 mg golimumab i henholdsvis uke 0 og 2, og deretter vedlikeholdsdoser på 50 mg eller 100 mg golimumab subkutant hver 4. uke hos UC -pasienter, nådde golimumabs serumkonsentrasjoner steady state omtrent 14 uker etter oppstart Behandling med golimumab 50 mg eller 100 mg subkutant hver 4. uke under vedlikehold resulterte i en gjennomsnittlig steady state gjennom serumkonsentrasjon på henholdsvis ca. 0,9 ± 0,5 mcg / ml og 1,8 ± 1,1 mcg / ml.
Hos UC -pasienter som ble behandlet med golimumab 50 mg eller 100 mg subkutant hver 4. uke, hadde samtidig bruk av immunmodulatorer ingen vesentlig effekt på steady state golimumab -nivåer.
Pasienter som utviklet antigolimumab-antistoffer hadde stort sett lave serum steady state-konsentrasjoner av golimumab (se pkt. 5.1).
Linjæritet
Golimumab, hos RA -pasienter, viste omtrent dose proporsjonale farmakokinetiske parametere over doseområdet 0,1 - 10,0 mg / kg etter en enkelt intravenøs dose Etter en enkelt SC dose hos friske personer Omtrentlige doseproporsjonale farmakokinetiske parametere ble også observert over doseområdet på 50 mg til 400 mg.
Effekt av vekt på farmakokinetikk
Det er en trend mot høyere tilsynelatende clearance for golimumab med vektøkning (se pkt.4.2).
Pediatrisk populasjon
Golimumabs farmakokinetikk ble bestemt hos 173 barn med pJIA mellom 2 og 17 år. I pJIA-studien hadde barn som ble behandlet med golimumab 30 mg / m2 (maks 50 mg) subkutant hver 4. uke median steady-state golimumab-lavkonsentrasjoner som var like på tvers av aldersgrupper og som også var like eller litt høyere enn de som ble sett hos voksne RA pasienter behandlet med golimumab 50 mg hver 4. uke.
Befolkningsfarmakokinetiske / farmakodynamiske modeller og simuleringer hos barn med pJIA bekreftet sammenhengen mellom golimumabs serumeksponering og klinisk effekt og støtter at golimumab 50 mg hver 4. uke doseringsregime hos barn med pJIA som veier minst 40 kg, gjør det mulig å oppnå eksponeringer som ligner dem som har vist seg å være effektive hos voksne.
05.3 Prekliniske sikkerhetsdata
Ikke-kliniske data viser ingen spesiell fare for mennesker basert på konvensjonelle studier av sikkerhetsfarmakologi, toksisitet ved gjentatt dosering, reproduksjon og utviklingstoksisitet.
Mutagenisitet, dyrs fruktbarhet eller langsiktige karsinogenisitetsstudier har ikke blitt utført med golimumab.
I en musefruktbarhet og generell reproduktiv funksjonsstudie ved bruk av et lignende antistoff som selektivt hemmer den funksjonelle aktiviteten til murint TNFa, ble antallet gravide mus redusert. Det er ikke kjent om disse resultatene skyldtes effektene hos hanner og / eller I en evolusjonær toksisitetsstudie utført på mus etter administrering av det samme analoge antistoffet og hos cynomolgus -aper som bruker golimumab, var det ingen indikasjon på mors toksisitet, embryotoksisitet eller teratogenisitet.
06.0 LEGEMIDDELOPPLYSNINGER
06.1 Hjelpestoffer
Sorbitol (E420)
L-histidin
L-histidinmonohydrokloridmonohydrat
Polysorbat 80
Vann til injeksjonsvæsker.
06.2 Uforlikelighet
I mangel av kompatibilitetsstudier, må dette legemidlet ikke blandes med andre produkter.
06.3 Gyldighetsperiode
22 måneder
06.4 Spesielle forholdsregler for lagring
Oppbevares i kjøleskap (2 ° C - 8 ° C).
Ikke frys.
Oppbevar den ferdigfylte pennen eller den ferdigfylte sprøyten i ytterkartongen for å beskytte mot lys.
06.5 Emballasje og innhold i pakningen
Simponi 50 mg injeksjonsvæske, oppløsning i ferdigfylt penn
0,5 ml oppløsning i en ferdigfylt sprøyte (type 1 glass) med en fast nål (rustfritt stål) og en kanylehett (gummi som inneholder latex), i en ferdigfylt penn. Simponi er tilgjengelig i pakninger som inneholder 1 ferdigfylt penn og flerpakninger som inneholder 3 (3 pakker med 1) ferdigfylte penner.
Simponi 50 mg injeksjonsvæske, oppløsning i ferdigfylt sprøyte
0,5 ml oppløsning i en ferdigfylt sprøyte (type 1 glass) med en fast nål (rustfritt stål) og en nålekappe (gummi som inneholder latex) Simponi er tilgjengelig i pakninger som inneholder 1 ferdigfylt sprøyte og flerpakninger som inneholder 3 (3 pakker med 1) ferdigfylte sprøyter.
Det er ikke sikkert at alle pakningsstørrelser blir markedsført.
06.6 Bruksanvisning og håndtering
Simponi leveres i en ferdigfylt penn til engangsbruk kalt SmartJect eller i en ferdigfylt sprøyte til engangsbruk.Hver Simponi -pakke kommer med bruksanvisning som beskriver hvordan du bruker pennen eller sprøyten. Når den ferdigfylte pennen eller den ferdigfylte sprøyten er fjernet fra kjøleskapet, bør den få romtemperatur ved å vente i 30 minutter før Simponi injiseres. Pennen eller sprøyten skal ikke ristes.
Løsningen er klar til svakt opaliserende, fargeløs til lysegul og kan inneholde noen små gjennomsiktige eller hvite proteinpartikler. Dette er ikke uvanlig for løsninger som inneholder proteiner.
Simponi må ikke brukes hvis løsningen har endret farge, er grumsete eller inneholder synlige fremmede partikler.
Fullstendige instruksjoner for klargjøring og administrering av Simponi i ferdigfylt penn eller ferdigfylt sprøyte finnes i pakningsvedlegget.
Ubrukte medisiner og avfall fra denne medisinen må kastes i henhold til lokale forskrifter.
07.0 INNEHAVER AV MARKEDSFØRINGSTILLATELSE
Janssen Biologics B.V.
Einsteinweg 101
2333 CB Leiden
Nederland
08.0 NUMMER FOR MARKEDSFØRINGSTILLATELSE
EU/1/09/546/001 1 ferdigfylt penn
EU/1/09/546/002 3 ferdigfylte penner
EU/1/09/546/003 1 ferdigfylt sprøyte
EU/1/09/546/004 3 ferdigfylte sprøyter
039541014
039541026
039541038
039541040
09.0 DATO FOR FØRSTE GODKJENNELSE ELLER FORNYELSE AV GODKJENNINGEN
Dato for første godkjenning: 1. oktober 2009
Dato for siste fornyelse: 19. juni 2014
10.0 DATO FOR REVISJON AV TEKSTEN
2. februar 2017
11.0 FOR RADIO -DRUGS, FULLSTÅENDE DATA OM DEN INTERNE RADIASJONSDOSIMETRI
12.0 FOR RADIOMEDLEM, YTTERLIGERE DETALJERTE INSTRUKSJONER OM EKSPORARISK FORBEREDELSE OG KVALITETSKONTROLL