Generellitet
Tuberøs sklerose er en genetisk sykdom som påvirker forskjellige organer og vev i menneskekroppen. Av denne grunn presenterer den et bredt spekter av symptomer, noen typiske for tidlig barndom, andre for voksen alder. Tuberøs sklerose kan overføres fra foreldre til barn, men det kan også oppstå på grunn av en spontan DNA -mutasjon.

Hva er tuberøs sklerose
Tuberøs sklerose er en genetisk lidelse preget av dannelse av hamartomi i forskjellige organer eller vev.
Hamartoma identifiserer et vevsområde hvor cellene har multiplisert seg ganske intenst og danner en merkbar masse, som ligner en klump eller knoll. Hamartomer minner om svulster, men bør ikke forveksles med dem: Faktisk er cellene i hamartoma identiske med de i vevet der de formerer seg; de på en svulst har derimot forskjellige egenskaper. Og gir øke til godartede neoplasmer, fibroider og angiofibromer.
Hjernen, huden, nyrene, øynene, hjertet og lungene er de mest berørte områdene, men de er ikke de eneste stedene. På grunn av mangfoldet av organer og vev som er involvert, er tuberøs sklerose også definert som en multisystem genetisk sykdom.
Senere vil det bli forstått hvorfor hamartomer bare vises i visse områder.
Epidemiologi
Dataene om forekomst og antall tilfeller over hele verden er usikre. Usikkerheten skyldes at mange pasienter ikke viser symptomer og lever et normalt liv.
Det er imidlertid anslått at forekomsten av tuberøs sklerose er ett tilfelle for hver 5000-10 000 nyfødte. Det er omtrent to millioner tilfeller verden over.
Det fører til
Tuberøs sklerose er en genetisk sykdom; dette betyr at et gen, som er tilstede i den berørte persons DNA, er mutert.
Genene som, når de påvirkes av de relative mutasjonene, forårsaker tuberøs sklerose er to:
- TSC1.
- TSC2.
Tilfellene av tuberøs sklerose observert så langt har bare ett av disse genene mutert. Derfor er den eneste mutasjonen av TSC1, eller av TSC2, tilstrekkelig til å forårsake tuberøs sklerose.
Studier utført i Europa og USA rapporterer at mutasjonen i TSC2 (80% av tilfellene) er mye hyppigere enn i TSC1 (de resterende 20%).
TSC1 OG TSC2
TSC1 -genet ligger på kromosom 9 og produserer et protein som kalles hamartin.
TSC2 -genet ligger på kromosom 19 og produserer et protein som kalles tuberin.
Proteinene som produseres, hamartin og tuberin, går sammen og fungerer sammen. Dette forklarer hvorfor mutasjonen av den ene eller den andre forårsaker den samme patologien.
FUNKSJON AV TSC1 OG TSC2
De regnes som svulstundertrykkende gener og spiller en grunnleggende rolle i prosessene for:
- Cellevekst og differensiering under embryogenese.
- Protein syntese.
- Autofagi.
Når TSC1 og TSC2 er mutert, er proteinene som produseres defekte, og disse fysiologiske prosessene finner ikke lenger sted regelmessig.
Cellevekst og differensiering under embryogenese
Protein syntese
Autofagi
Cellevekst og differensiering under embryogenese
Protein syntese
Autofagi
ONSET OF AMARTOMAS
Hamartomer kan oppstå når en mutasjon oppstår i et gen som styrer cellevekst og differensiering, for eksempel TSC1 eller TSC2. Cellene vokser følgelig i antall og genererer tydelige masser; på denne måten dannes det plaketter som ligner en knute eller en knoll i form. I histologi er denne prosessen definert av begrepet hyperplasi.
GENETIKK
To premisser:
- Hvert menneskelig DNA -gen er tilstede i to kopier. Slike kopier kalles alleler.
- Mennesket har 23 par kromosomer. Av disse er det bare ett par som bestemmer kjønn (kjønnskromosomer), alle de andre kalles autosomale kromosomer.
Tuberøs sklerose er en autosomal genetisk sykdom dominerende. For dette er det tilstrekkelig at en allel er mutert for at hele genet ikke skal fungere skikkelig. Den muterte allelen har faktisk mer kraft enn den friske (dominans).
Faktisk forverres tuberøs sklerose når både TSC1 eller TSC2, alleler er mutert. Med andre ord gir ikke bare en allel, selv om den er dominant over den andre, ingen åpenbare symptomer.I disse tilfellene snakker vi om alleler med ufullstendig dominans.
ARV € ELLER SPONTAN MUTASJON?
TSC1, eller TSC2, mutasjon kan oppstå fra:
- Arvelig overføring (dvs. fra en av de to foreldrene) av en mutert allel.
- Spontan mutasjon av en allel i embryonalt stadium (eller embryogenese).
En tredjedel av tuberøs sklerose tilfeller skyldes arvelig overføring. I disse tilfellene er det nok at en forelder har en mutasjon av TSC1- eller TSC2 -genene for at avkomene skal bli påvirket av sykdommen (vi har faktisk sett at tuberøs sklerose er en autosomal dominerende arvelig sykdom).
De resterende 2/3 tilfellene skyldes en spontan mutasjon under embryonalt stadium.
TSC1 på 50%
TSC2 i de resterende 50%
TSC2 i 70%
TSC1 i 30%
HVORFOR BLIR BARE SIKKERE ORGANER?
Premiss: embryoet, i de første stadiene av utviklingen, har tre lag med celler:
- Ectoderm, den ytterste.
- Mesoderm, det sentrale.
- Endoderm, den innerste.
Spesifikke organer og vev stammer fra hvert lag.
Nervesystemet
Epidermis
Epitel i munnen
Epitel i tykktarmen
Kåt og krystallinsk
Tannemalje
Hudbein
Hjerte
Nyre
Tarmveggforing
Muskulatur i lemmer
Serøse membraner i lungene (pleura) og hjertet (perikard).
Lever
Bukspyttkjertelen
Fordøyelsessystemet
Vi har nå alle elementene for å forstå hvorfor hamartomer bare oppstår i visse deler av kroppen.
Mutasjoner av TSC1 eller TSC2 forekommer på embryonalt stadium i cellene i ektoderm og mesoderm. Derfor vil vevet, som vil oppstå fra disse cellelagene, presentere hamartomer.
Symptomer
For ytterligere informasjon: Tuberøs sklerose - årsaker og symptomer
Organer og vev som påvirkes av tuberøs sklerose er mange. Distriktene som er mest berørt er:
- Hjerne, hud, nyrer, hjerte, øyne
Men andre, sjeldnere plager bør ikke glemmes, til skade for:
- Lunger, tarm, lever, tenner, endokrine system, bein
Noen symptomer vises i ung alder, andre i voksen alder.
UFULLT DOMINANS
Det har allerede blitt nevnt ovenfor at dominansen av den muterte allelen til TSC1- eller TSC2 -genene er ufullstendig. Dette betyr at den friske allelen fremdeles er i stand til å produsere et "sunt" protein (hamartin eller tuberin), om enn i lavere mengde. Tilstedeværelsen av det "sunne" proteinet kompenserer for skaden forårsaket av det muterte proteinet. Under disse forholdene forårsaker hamartomer ennå ikke dramatiske manifestasjoner.
Når den andre allelen også endres (dette er en sjelden hendelse, men mulig), vokser hamartomene på en ukontrollert måte.
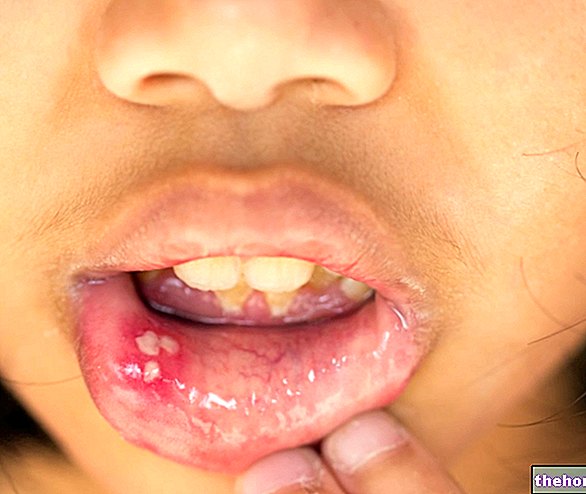
HUD MANIFESTASJONER
Omtrent 90% av pasientene har hudforandringer. Arrangementene er mange og varierte. Typiske er depigmenterte flekker, Pringles sebaceous adenomer og Koenen neglesvulster.
Depigmenterte flekker er hypomelanotiske flekker, det vil si med et lavere melanininnhold
Pringle sebaceous adenomer er godartede svulster, også kalt ansiktsangiofibromer. Hamartomas fremstår som små, kuleformede, knallrøde masser. Koenen neglesvulster er fibroider og stammer fra hamartomer på noen få millimeter.
Bilder på hud manifestasjoner av tuberøs sklerose
Tabellen viser de mange hudmanifestasjonene på grunn av tuberøs sklerose:
Stamme
Kunst
Kinn
Nese
Hake
Negler og hender
Front
Hodebunnen din
Stamme
Dorsal-lumbalregionen
Nakke
Skulder
Tenner
Munn
Fremre tannkjøtt
Leppe
Gane
NEUROLOGISKE SYMPTOMER
Hjernestedene som er påvirket av tuberøs sklerose er:
- Hjernebarken
- Den hvite saken
- Ventriklene
- De basale ganglier
De to figurene hjelper leseren med å forstå de berørte områdene.

Avhengig av hamartomas beliggenhet og form, kan det oppstå forskjellige lidelser, for eksempel:
- Epilepsi
- Subependymale knuter
- Hjernesvulster av typen astrocytom
- Psykiske, atferds- og læringsunderskudd.
Knoll
Bark
80-90%
- Kramper
- Delvis
- Feberlig
Svært tidlig barndom (spasmer), 75%
Voksen alder (delvis), 25%
Nodule
Ventrikler
80-90%
Barndom
Obstruktiv hydrocephalus
Evolusjon til subpendimal astrocytom
Hjernecyster
Nodule
> 1 cm
Ventrikler (Foramina di Monro)
6%
Mellom 4 og 10 år
Hodepine
Han retched
Kramper
Endringer i synsfeltet
Plutselige humørsvingninger
Hydrocephalus
Hjernecyster
Psykisk handikap
Tidlig barndom
(0-5 år)
Krever tilsyn (85%)
Fravær av språk (65%)
Ikke selvforsynt (60%)
Autisme
Oppmerksomhetsunderskudd
Hyperaktivitet
Aggresjon
Selvlemlestelse
Søvnforstyrrelser
Barndom
Forening med epilepsi
Vanskelig familie- og skoleledelse
KINNESKADER
De er veldig hyppige. Faktisk vises de i 60-80% av tilfellene. De består av:
- Hamartomas som ligner godartede svulster.
- Misdannelser i nyrestrukturen.
Angiomyolipom (60-70%)
Angiolipom
Myolipomer
De er godartede svulster, som vises i flere former
I barndommen: Asymptomatisk
I voksen alder: Mulig ruptur av hamartoma, etterfulgt av blødning, hematuri og magesmerter.
Nyresvikt
Hestesko nyre
Polycystisk nyre
Mangel på nyre (renal agenese)
Dobbel urinleder
KARDIOVASKULÆRE SKADER
Igjen skyldes de hamartomer som ligner godartede svulster, kalt rabdomyomer.
Asymptomatisk.
Hvis dimensjonene er store:Arytmier
Endringer i hjerteflyten
LUNG SKADER
De skyldes hovedsakelig pulmonal lymfangioleiomyomatose (AML) og i mindre grad mikronodulær multifokal hyperplasi. De er typiske manifestasjoner av voksen alder.
Sjelden sykdom
Det rammer hovedsakelig voksne kvinner
Lungecyster vises
De fleste tilfeller er asymptomatiske
Symptomer er: astmalignende dyspné, hoste, spontan pneumotoraks, respirasjonssvikt
Sjelden sykdom
Det rammer hovedsakelig voksne, menn og kvinner
Nodler vises, synlige på et røntgenbilde av brystet
Nesten alltid asymptomatisk
ANDRE SKADER
Retinal hamartom
Retinal astrocytom
Tarmpolypper
Tarmcyster
Angiomyolipom
Angiomer
Pseudo-cyste i hender og føtter
Adenomer
Angiomyolipomer
Diagnose
Diagnosen består av:
- Anamnese
- Klinisk analyse av de nevnte tegnene
- Instrumentale undersøkelser
ANAMNESIS
Legen gjør en "undersøkelse av pasientens familiehistorie for å forstå om tuberøs sklerose er arvet eller på grunn av en spontan mutasjon.
KLINISK ANALYSE AV SKILTENE
I 1998 etablerte en gruppe internasjonale leger et diagnostisk kriterium basert på de nevnte kliniske manifestasjonene. De har blitt delt inn i:
- Store tegn (eller kriterier)
- Mindre tegn (eller kriterier)
Hvis pasienten viser
- 2 store tegn,
- 1 større og 2 mindre tegn
Hvis pasienten viser
- 1 hovedtegn
- 2 eller flere mindre tegn
Klassifiseringen av skiltene er som følger:
INSTRUMENTALE UNDERSØKELSER
CT -skanning av hjernen
Kjernemagnetisk resonans
- Knoller i hjernebarken
- Subependymale knuter
- Giant celle subependymale astrocytomer (SEGA)
Ja (ioniserende stråling)
Nei
Spirometri
Røntgen av brystet
- Lungelymfangioleiomyomatose
- Respirasjonssvikt
Nei
Ja (ioniserende stråling)
GENETISK TEST
Det er en lang undersøkelse som tar et par måneder. Den er derfor ikke nyttig for tidlig diagnose, men snarere en bekreftelse av diagnosen basert på kliniske tegn.
Terapi
Det er ingen spesifikk og effektiv kur, ettersom tuberøs sklerose er en:
- Genetisk sykdom.
- Multisystem sykdom.
Noen symptomer kan imidlertid dempes for å unngå komplikasjoner og forbedre pasienters livskvalitet.
FARMAKOLOGISK BEHANDLING
De kliniske manifestasjonene som kan behandles med administrering av legemidler er:
- Infantil epilepsi
- Lungelymfangioleiomyomatose (LAM)
- Nyreplager
Infantil epilepsi. Den lille pasienten får krampestillende medisiner:
- ACTH (adrenokortikotropt hormon)
- Vigabatrin
Lungelymfangioleiomyomatose. Bronkodilatatorer av beta-2-agonisttypen, slik som salbutamol, er nyttige. Imidlertid er effekten av hormonbehandling basert på progesteron eller buserelin usikker
Nyreplager. Antihypertensiva, som ACE -hemmere og diuretika, brukes.
FYSISK-KIRURGISKE BEHANDLINGER
De består av tiltak som tar sikte på å fjerne:
- Ansiktsangiofibromer
- Spiker fibroids
- Hudplakkene
- De riflede flekkene
- Giant celle subependymale astrocytomer (SEGA)
- Nyre angiomyolipomer
- Lungeskader
- Knollene i hjernebarken, som forårsaker epilepsi
Tabellen nedenfor oppsummerer de viktigste terapeutiske behandlingene og deres egenskaper.
Diatermi
Kryoterapi
Kirurgisk fjerning
Minimalt invasiv
Jepp
Laserterapi
Kirurgisk fjerning
Jepp
Oppfølging og prognose
Innledning: det medisinske uttrykket oppfølging refererer til pasienten som, som lider av kreft, har blitt positivt operert.
Periodiske kontroller anbefales for oppfølging. Oftalmoskopi, dvs. fundusundersøkelse, kan også utføres en gang i året, men omvendt krever nevrologiske, hjerte- og nyreforhold hyppigere overvåking.
PROGNOSE
Utviklingen av tuberøs sklerose er variabel og avhenger fra sak til sak.
Noen pasienter viser milde, nesten umerkelige symptomer. For disse påvirkes ikke livskvaliteten av sykdommen og prognosen er utmerket.
Omvendt viser andre pasienter mye mer dramatiske og tydelige symptomer. Døden skjer hovedsakelig på grunn av nevrologiske lesjoner, derfor blir prognosen svært ugunstig.
GENETISK KONSULTERING
Hvis en av foreldrene har tuberøs sklerose, er sannsynligheten for at et barn arver den samme tilstanden 50%.
Hvis et barn av friske foreldre derimot påvirkes, er sannsynligheten for at et andre barn blir syk, veldig lav. I disse tilfellene avklarer en genetisk test om foreldrene er bærere av tuberøs sklerose, eller om det i stedet har skjedd en spontan mutasjon.

















.jpg)