På grunn av mutasjonen av NSD1 -genet er Sotos syndrom en tilstand som er oppnådd under embryonal utvikling, i mer enn 90% av tilfellene, og en arvelig tilstand, i prosentandelen av gjenværende tilfeller; Derfor er det nesten alltid et resultat av en ny mutasjonshendelse.
Symptomene og tegnene på Sotos syndrom kan delvis sees allerede ved fødselen, noe som gjør tidlig diagnose mulig.
Dessverre kan de som lider av Sotos syndrom for tiden bare stole på symptomatiske behandlinger - det vil si rettet mot å lindre symptomene - ettersom det ikke finnes noen kur som kan avbryte konsekvensene av mutasjonen i NSD1 -genet.
Andre navn på Sotos syndrom
Sotos syndrom er også kjent som hjernegigantisme (refererer til) eller Sotos-Dodge syndrom.
Epidemiologi
Ifølge statistikk er en av hver 10.000-14.000 individer født med Sotos syndrom.
befruktet egget og embryogenesen begynte) og, bare i de resterende 5-10% av kliniske tilfeller, arvelig (dvs. overført via foreldrerute).Dermed er Sotos syndrom i 90-95 tilfeller av 100 en tilstand som skyldes en "ny" mutasjon, og i bare 5-10 tilfeller av 100 er en tilstand arvet fra foreldrene.
Hva forårsaker genmutasjonen forbundet med Sotos syndrom?
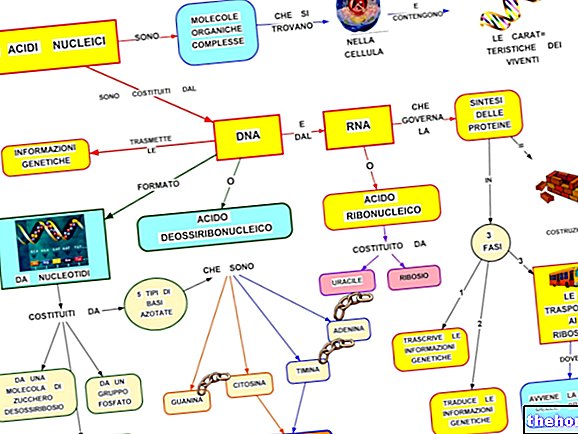
Premiss: Genene på menneskelige kromosomer er DNA -sekvenser som har til oppgave å produsere grunnleggende proteiner i biologiske prosesser som er viktige for livet, inkludert cellevekst og replikasjon.
I fravær av mutasjoner i ladningen produserer NSD1 -genet et protein fra gruppen av histonmetyltransferaser, hvis funksjon er å regulere aktiviteten til gener som er involvert i prosessene for vekst og utvikling, og unngå cellulære hyperproliferasjonsfenomener.
Mer enkelt…
Hos friske mennesker produserer NSD1 et protein som fint kontrollerer aktiviteten til en rekke gener som er involvert i veksten og utviklingen av menneskekroppen.
I nærvær av mutasjonen som er ansvarlig for Sotos syndrom, mister NSD1 -genet en del av sin regulatoriske kapasitet mot de nevnte genene (NB: tapet skyldes en kvantitativ nedgang i NSD1), og dette fører til et "unormalt uttrykk for sistnevnte" med konsekvenser for kropp og mental utvikling.
Mer enkelt…
Hvis den er mutert, er NSD1 lav, og dette innebærer en "anomal aktivitet fra genene under dens kontroll, som igjen følger en" endring av fysisk og mental utvikling.
Sotos syndrom er en autosomal dominerende sykdom
Å forstå...
Hvert menneskelig gen er tilstede i to kopier, kalt alleler, en av mors opprinnelse og en av farlig opprinnelse.
Sotos syndrom har alle egenskapene til en autosomal dominerende sykdom.
En genetisk sykdom er autosomaldominant når mutasjonen av en enkelt kopi av genet som forårsaker det er tilstrekkelig til å manifestere seg.
Til dette er det viktig å legge til at virkningen av Sotos syndrom på skjelettvekst også påvirker beinaldring: sammenlignet med sine friske jevnaldrende, viser barn med Sotos syndrom en beinalder som er 2-4 år høyere.
HAR STOR VEKST SLUTT?
De viktigste effektene på skjelettvekst ved Sotos syndrom slutter rundt 3-4 års levetid. Fra denne alderen og utover har den høye graden av skjelettvekst en gradvis tendens til å avta opp til normalitet.
KONSEKVENSER I VOKSENALDER
På grunn av overdreven skjelettvekst i en veldig ung alder, når personer med Sotos syndrom i voksen alder en høyde på ikke mindre enn 193 cm.
Det er interessant å påpeke for leserne at vekten til disse fagene er i tråd med vekten til friske individer med samme høyde; dette betyr at Sotos syndrom påvirker bare høyden og ikke kroppsvekten.
Kraniofaciale anomalier
Kraniofaciale anomalier relatert til Sotos syndrom er:
- Fremtredende panne;
- Avtagende hårfeste
- Dolichocephaly (dvs. langt, smalt hode);
- Unormalt brede øyne (okulær hypertelorisme);
- Spiss hake;
- Øyelokkspalter som peker nedover;
- Smal gane;
- Langt og smalt ansikt.
De ovennevnte avvikene er spesielt tydelige ved fødselen; da, med vekst, har de en tendens til å "visne", for å være mindre synlige. De eneste stabile kraniofaciale tegnene selv i voksen alder er: fremtredende hake, dolichocephaly, fremtredende panne og avtagende hårfeste.
Forsinkelse i intellektuell utvikling
Ved Sotos syndrom er forsinkelsen i den intellektuelle utviklingen en konsekvens av et problem i sentralnervesystemet.
For å avsløre denne intellektuelle forsinkelsen er: en viss treghet i å lære å snakke, språkproblemer (f.eks. Stamme, problemer med lydgjengivelse, monoton stemme, etc.) og redusert IQ (stabil for livet).
Visste du at ...
For 80-85% av pasientene med Sotos syndrom er IQ litt over 70 (derfor 30-40 poeng under normalen); for den resterende prosentandelen er det imidlertid normalt.
Forsinkelse i motorisk utvikling
Ved Sotos syndrom manifesteres forsinket motorisk utvikling av:
- Vanskelighetsgrad å gå på alle fire (det er helt klart et problem i veldig ung alder);
- Muskelhypotoni;
- Klønete og koordineringsvansker (for eksempel er det vanskelig å lære å sykle);
- Motoriske problemer på grunn av dårlig kontroll over de stripete musklene.
Med unntak av den første har alle de andre manifestasjonene nevnt ovenfor en tendens til å opprettholdes gjennom livet.
Atferdsproblemer

De typiske atferdsproblemene som kan sees hos pasienter med Sotos syndrom er: Attention Deficit Hyperactivity Disorder (ADHD), ulike typer fobier, aggresjon, impulsivitet, irritabilitet og tvangslidelser.
Andre symptomer
Noen ganger, til symptomatologien som er rapportert ovenfor - som representerer en klassisk symptomatologi - kan Sotos syndrom legge til en rekke andre viktige kliniske konsekvenser, som er:
- Gulsott (spesielt ved fødselen)
- Manglende evne til å mate (spesielt ved fødselen)
- Epilepsi;
- Tap av hørsel;
- Skoliose (hos omtrent 40% av pasientene);
- Misdannelser i nyrene og / eller urinveiene (hos omtrent 20% av pasientene);
- Hjertefeil (hos ikke mer enn 35% av pasientene);
- Visjonsproblemer, som for eksempel å myse
- Disposisjon for utvikling av luftveisinfeksjoner;
- Disposisjon for utvikling av ondartede svulster.
Medisinsk historie og fysisk undersøkelse
Anamnese og fysisk undersøkelse består hovedsakelig i en nøyaktig evaluering av symptomene som pasienten viser.
I en kontekst av Sotos syndrom er det i disse fasene av diagnoseprosessen at legen fastslår tilstedeværelsen av kraniale og ansiktsanomalier, unormal skjelettvekst, svekket intellektuell utvikling, forsinkelse i motorisk utvikling og andre mindre vanlige manifestasjoner.
Radiologiske undersøkelser
Radiologiske undersøkelser brukes til å oppdage og analysere eventuelle abnormiteter som kan påvirke urinveiene, hjertet eller ryggraden (skoliose).
Genetisk test
Det er DNA -analysen som tar sikte på å påvise mutasjoner i kritiske gener.
I sammenheng med Sotos syndrom representerer den den bekreftende diagnostiske testen, da den tillater å markere NSD1 -mutasjonen.
Når stilles diagnosen?
Sotos syndrom kan diagnostiseres fra fødselen.
Det skal imidlertid bemerkes at ingen av de rutinemessige undersøkelsene, utført i prenatalfasen, tillater oss å identifisere den aktuelle sykdommen; for prenatal diagnose av Sotos syndrom, er det faktisk nødvendig med en prenatal genetisk test (etter en "fostervannsprøve eller CVS).

- Logoterapi (eller språkterapi) for å dempe språkforstyrrelser;
- Fysioterapi for å lindre bevegelsesproblemer;
- En medikamentell terapi ad hoc, rettet mot å håndtere ADHD (hvis noen) og / eller andre atferdsproblemer;
- Bruk av korrigerende briller (hvis det er synsproblemer);
- Farmakologisk eller kirurgisk terapi fokusert på å kontrollere hjerte- og / eller urinveisproblemer;
- Implementering av et program av screening onkologisk, rettet mot rettidig identifisering av alle ondartede svulster (husk at Sotos syndrom favoriserer neoplastiske prosesser).
Hvilke medisinske tall innebærer symptomatisk behandling av Sotos syndrom?
Symptomatisk behandling av Sotos syndrom krever koordinert intervensjon fra flere medisinske spesialister, inkludert: barneleger, genetikere, endokrinologer, nevrologer, logopeder, ortopediske kirurger, øyeleger, psykiatere og fysioterapeuter.