Generellitet
Osteogenesis imperfecta er en medfødt genetisk sykdom, som ikke er knyttet til sex, som er ansvarlig for en viss beinskjørhet og en markert tendens til brudd.
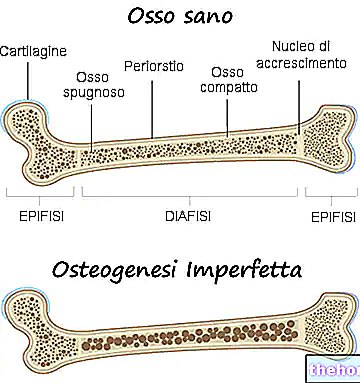
Symptomene på osteogenesis imperfecta er mange; generelt består de av: bein svekkelse, høy tendens til beinbrudd, tilstedeværelse av blå, grå eller lilla okulære sclerae, tilstedeværelse av bein deformiteter eller andre skjelettforandringer, trekantet ansikt, tannskjørhet, etc. .
Generelt er følgende avgjørende for en korrekt diagnose av osteogenesis imperfecta: fysisk undersøkelse, medisinsk historie, medisinske avbildningstester, en type I kollagenvurderingstest og en genetisk test.
Dessverre er de eneste behandlingene som er tilgjengelige for pasienter med osteogenesis imperfecta for tiden symptomatiske. Den aktuelle sykdommen er faktisk uhelbredelig.
Hva er osteogenesis imperfecta?
Osteogenesis imperfecta er en genetisk sykdom som gjør den berørte persons bein svakere og mer utsatt for brudd.
I virkeligheten, med begrepet osteogenesis imperfecta, refererer leger til en heterogen gruppe genetiske sykdommer, preget av en viss grad av beinskjørhet. Det er derfor flere former (eller typer) for osteogenesis imperfecta, noen mye mer alvorlige enn andre.
DET ER EN KONGENITAL SYKDOM
Hos mennesker som er rammet av det, er osteogenesis imperfecta en sykdom som er tilstede fra fødselen, og derfor kan den for all del definere en medfødt sykdom.
ER DET SEKSRELATERT?
Osteogenesis imperfecta er ikke en kjønnsrelatert genetisk sykdom, for eksempel hemofili eller Klinefelters syndrom.
EPIDEMILOGI
Ifølge noen statistiske undersøkelser vil forekomsten av osteogenesis imperfecta være lik et tilfelle hver 15. 000-20.000 fødsler. Dette betyr at hver 15.000-20.000 nyfødte får en som er påvirket av osteogenesis imperfecta.
Andre statistiske studier har også vist at osteogenesis imperfecta påvirker menn og kvinner likt, og at det ikke foretrekker en bestemt befolkning eller etnisk gruppe.
Levetid er en ekstremt variabel parameter, som avhenger av formen for osteogenesis imperfecta.
Årsaker
Osteogenesis imperfecta skyldes nesten alltid en kvalitativ og kvantitativ endring av produksjonen av type I kollagen.
Type I kollagen er avgjørende for å styrke bein og for å opprettholde friske bindevev som utgjør brusk, sener, hud, okulær sclera, etc.
Derfor påvirker en endring i produksjonen av type I kollagen styrken til beinene og god helse til bindevevet som er tilstede i menneskekroppen.
HVA ENDRER COLLAGEN -PRODUKSJONEN?
En genetisk sykdom er en tilstand som oppstår på grunn av en mutasjon av ett eller flere gener som utgjør cellulært DNA.
Når det gjelder osteogenesis imperfecta, er årsakene til sistnevnte nesten alltid å finne i mutasjonen av en eller begge genene COL1A1 (lokalisert på kromosom 17) og COL1A2 (lokalisert på kromosom 7).
Under normale forhold regulerer COL1A1 og COL1A2 normal produksjon av type I kollagen; i nærvær av mutasjoner i ladningen, mislykkes de i deres regulatoriske funksjon.
Viktig: Hvilke andre gener, hvis de er mutert, forårsaker osteogenesis imperfecta?
I tillegg til mutasjonene av COL1A1 og COL1A2, er mutasjoner i IFITM5, SERPINF1, CRTAP og LEPRE1 gener potensielle årsaker til osteogenese imperfecta.
De nevnte genene dekker funksjoner som er forskjellige fra COL1A1 og COL1A2 - derfor kontrollerer de ikke produksjonen av kollagen av type I - men de har fortsatt en "innflytelse på styrken og motstanden i beinene i det menneskelige skjelettet.
HVILKEN GENETISK SYKDOM ER DET?
Osteogenesis imperfecta er en autosomal genetisk sykdom.
Begrepet autosomal, assosiert med en genetisk sykdom, indikerer at den aktuelle tilstanden skyldes genetiske mutasjoner basert på autosomale og ikke-kjønnskromosomer.
Leserne blir minnet om at mennesket har et kromosomalt sett med 23 par totale kromosomer, hvor 22 par er av autosomaltypen og bare ett par er av seksuell type. Paret kromosomer av den seksuelle typen påvirker kjønnet til den individ.
Osteogenesis imperfecta etter mutasjoner i COL1A1, COL1A2 og IFITM5 har alle egenskapene til en autosomal dominerende sykdom.Når det skyldes mutasjoner i genene SERPINF1, CRTAP og LEPRE1, har det egenskapene til en autosomal recessiv sykdom.
TYPER
For tiden tror leger at det er 8 typer (eller former) for osteogenesis imperfecta. For å skille de forskjellige typene bestemte de seg for å bruke den romerske nummereringen, for å være presis de første åtte romertallene.
Tabellen nedenfor viser de 8 formene for osteogenesis imperfecta, mutasjonene som forårsaker dem og andre egenskaper.
Fyr
Mutert gen
Type genetisk sykdom
DE
COL1A1
Autosomal dominerende
II
COL1A1 og COL1A2
Autosomal dominerende
III
COL1A1 og COL1A2
Autosomal dominerende
IV
COL1A1 og COL1A2
Autosomal dominerende
V.
IFITM5
Autosomal dominerende
DU
SERPINF1
Autosomal recessiv
VII
CRTAP
Autosomal recessiv
VIII
HARE 1
Autosomal recessiv
* NB: åpenbart er mutasjonene i COL1A1 og COL1A2, som forårsaker de fire første formene for osteogenesis imperfecta, genetiske endringer med litt forskjellige egenskaper. Ellers hadde det ingen mening å skille det ene fra det andre.
Symptomer, tegn og komplikasjoner
Alle typer osteogenesis imperfecta er ansvarlige for en svekkelse av beinene, slik at personen som er rammet av sykdommen har en spesiell disposisjon for brudd. Graden av svekkelse av beinene varierer etter formen; for noen av disse er denne svekkelsen større enn for andre.
Når det er sagt, må det påpekes at hver form for osteogenesis imperfecta har sitt eget symptomatiske bilde, som for noen kan huske det symptomatologiske bildet av andre former.
MULIGE symptomer og tegn
De mulige symptomene og tegnene på osteogenesis imperfecta inkluderer:
- Tilstedeværelse av beinmisdannelser;
- Tilstedeværelse av en kort og liten kropp (beregnet som bagasjerommet);
- Leddproblemer (f.eks. Løse ledd);
- Muskel svakhet;
- Blå, lilla eller grå okulær sclera;
- Trekantet ansikt;
- Fat bryst;
- Morfologiske anomalier i ryggraden;
- Tannskjørhet;
- Nedgang eller totalt tap av hørsel;
- Pusteproblemer
- Problemer knyttet til fravær eller mangel på type 1 kollagen.
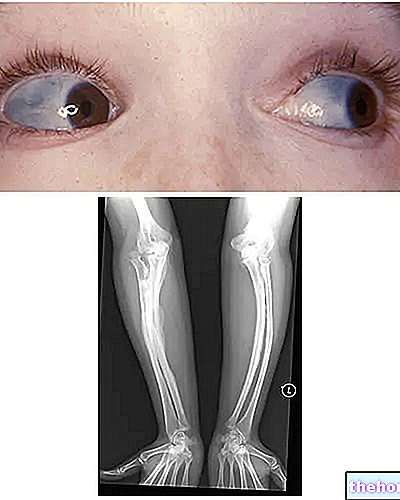
Osteogenesis Imperfecta: legg merke til blåfargen på sclerae og beindeformasjonene som kjennetegner sykdommen. Fra wikipedia.org
HVA ER DE MEST ALVORLIGE FORMENE FOR IMPERFEKT OSTEOGENESIS?
Leger klassifiserer den symptomatologiske alvorlighetsgraden av de forskjellige typene osteogenesis imperfecta på en skala på 3 grader, som er: den milde graden, den moderate graden og den alvorlige graden.
Bare én form tilhører kategorien "mild grad": "type I osteogenesis imperfecta"; 4 former for osteogenesis imperfecta tilhører kategorien "moderat grad": IV, V og VI; til slutt tilhører kategorien "alvorlig grad" 3 former: II, III, VII og VIII.
TYPE I: FUNKSJONER
Den vanligste og minst alvorlige formen av alle, type I osteogenesis imperfecta har følgende egenskaper:
- Det forårsaker brudd spesielt før puberteten;
- Det har "nesten ingen innflytelse på høyden, så pasientene er vanligvis av normal" høyde;
- Forårsaker leddproblemer og muskelsvakhet
- Det er ansvarlig for blå, lilla eller grå sclera;
- Det er årsaken til trekantet ansikt og ryggmargsavvik;
- Det forårsaker nesten aldri bein deformiteter. Hvis det provoserer dem, er de minimale;
- Det kan forårsake tannskjørhet og / eller hørselstap (sistnevnte oppstår vanligvis i voksen alder);
- Det er assosiert med tilstedeværelsen av type I kollagen som er normal i kvalitet, men unormalt i mengde (det er dårligere enn normalt).
TYPE II: FUNKSJONER
Type II osteogenesis imperfecta er preget av:
- Dødsårsak ved fødsel eller kort tid etter. Åndedrettsproblemer forårsaker nesten alltid død;
- Tilstedeværelse av betydelig beinskjørhet og alvorlige beindeformiteter;
- Kort vekst og underutviklede lunger
- Blå, lilla eller gråfarget sclera;
- Tilstedeværelse av kvantitative og kvalitative anomalier av type I kollagen.
TYPE III: FUNKSJONER
Type III osteogenesis imperfecta har følgende egenskaper:
- Selv om det er veldig alvorlig, forårsaker det ikke ofte død i den nyfødte perioden;
- Det er assosiert med "høy skjørhet i bein;
- Det er ansvarlig for kort statur, leddproblemer, muskelsvakhet (spesielt i bena og armene), fatbryst, trekantet ansikt og unormal krumning av ryggraden;
- Det skyldes blå, lilla eller grå sclera;
- Det kan forårsake pusteproblemer, tannsprøhet og hørselstap;
- Det er ofte ansvarlig for beindeformiteter;
- Det er assosiert med kvalitative og kvantitative abnormiteter av type I kollagen.
TYPE IV: FUNKSJONER
Type IV osteogenese er preget av:
- En grad av skjørhet i bein mellom form II og III og form I;
- Kortere enn gjennomsnittlig vekst;
- Blå, lilla eller gråfarget sclera;
- Bendeformiteter av mild / moderat enhet, små abnormiteter i ryggraden og fatbrystet;
- Trekantet ansikt;
- Mulig tilstedeværelse av tannskjørhet og hørselstap;
- Tilstedeværelse av type I kollagenavvik.
TYPE V: FUNKSJONER
Type V osteogenesis imperfecta ligner på noen måter osteogenesis imperfecta av type IV. Den har imidlertid noen særegenheter, som er:
- Normal farget sclera;
- Fravær av dental skjørhet;
- Dannelse av unormale benete calluses, under helingsprosessen av brudd i bein;
- Forkalkning av den interosseøse membranen som befinner seg mellom radius og ulna. Dette svekker underarmens mobilitet.
TYPE VI: FUNKSJONER
Også type VI osteogenesis imperfecta ligner på form IV. For å skille den fra sistnevnte er noen særegenheter, inkludert de høye nivåene av alkalisk fosfatase i blodet og tilstedeværelsen på noen bein av lameller (benete) som ligner på pigger av fisk.
TYPE VII: FUNKSJONER
Symptomatisk kan osteogenese imperfecta type VII i noen tilfeller ligne type IV og type II i andre tilfeller.
Det særegne ved denne alvorlige patologiske formen inkluderer:
- Kortvokst;
- Tilstedeværelsen av en ekstremt kort humerus (armben) og lårbenet (lårbenet);
- Den hyppige tilstedeværelsen av en hofteformitet, kjent som coxa vara.
TYPE VIII: EGENSKAPER
Type VIII osteogenesis imperfecta minner veldig om form II og III.
Blant de særegne egenskapene skiller følgende seg ut: det alvorlige vekstunderskuddet, den alvorlige skjeletthypomineraliseringen og fraværet (eller mangelen på tilstedeværelse) av prolyl 3-hydroksylase-enzymet.
Diagnose
Generelt begynner den diagnostiske prosessen som pasienter med en mistenkt form for osteogenese imperfecta blir utsatt for, med en grundig fysisk undersøkelse og en forsiktig sykehistorie; så fortsetter den, med en "analyse av pasientens familiehistorie og med en serie diagnostiske bildetester (røntgenstråler, CT-skanninger, etc.); til slutt ender den med en kvantitativ og kvalitativ evaluering av type I kollagen og med en genetisk test.
I dag er det mulighet for å diagnostisere osteogenesis imperfecta selv i prenatalfasen, ved å utsette en gravid kvinne for ultralyd.
VIKTIGHETEN FOR MÅL EKSAMEN OG HISTORIEN
En legeekspert i osteogenesis imperfecta er i stand til, veldig ofte, å diagnostisere den nevnte sykdommen, bare bare ved hjelp av fysisk undersøkelse og anamnese. Dette betyr at disse diagnostiske testene ikke har noen ubetydelig betydning.
EVALUERING AV TYPE I COLLAGEN -PRODUKSJON
Som regel er den kvalitative og kvantitative evalueringen av type I kollagen en veldig pålitelig test, siden de fleste tilfeller av osteogenesis imperfecta som nevnt er preget av mutasjoner i genene som styrer produksjonen av type 1 kollagen.
For å vurdere mengden og kvaliteten på type I kollagen som er tilstede på mobilnivå hos en person, kan leger stole på en hudbiopsi eller en bestemt blodprøve.
Begge disse evalueringstestene er ganske komplekse, og pasienten (eller foreldrene) må kanskje vente flere uker for å kjenne resultatene.
GENETISK TEST
Gjennom en genetisk test som undersøker hele DNA -en til individet som undersøkes, kan leger definitivt bestemme egenskapene til den tilstedeværende genetiske mutasjonen.
Vanligvis er det utført en genetisk test på alt cellulært DNA når evalueringen av egenskapene til type I -kollagen ikke har gitt de ønskede resultatene, eller når det ikke er en mutasjon i COL1A1 eller COL1A2 som forårsaker "osteogenesis imperfecta".
PRENATAL DIAGNOSE
Prenatal ultralyd er veldig nyttig for å identifisere type II og type III osteogenesis imperfecta.
Terapi
Det er foreløpig ingen spesifikk kur mot osteogenesis imperfecta. Med andre ord er mennesker med osteogenesis imperfecta bestemt til å leve med den nevnte tilstanden til døden, noe som ofte skyldes konsekvensene av selve sykdommen.
Mangelen på spesifikk terapi utelukker ikke eksistensen av andre behandlingsformer. Faktisk, blant de terapeutiske mulighetene til en pasient med osteogenesis imperfecta, er ulike symptomatiske terapier inkludert; Med symptomatisk behandling mener vi behandlinger som er i stand til å lindre symptomer, bremse sykdomsforløpet og forhindre (eller i det minste utsette) de alvorligste konsekvensene.
MULIG SYMPTOMATISKE BEHANDLINGER
I listen over mulige symptomatiske behandlinger for osteogenesis imperfecta skiller følgende seg ut:
- Kirurgisk innsetting, inne i de lengste beinene (N.B: den mest utsatte for brudd), av negler som gir større motstand mot brudd og deformiteter. Denne operasjonen kalles rodding intramedullary;
- Konservativ eller kirurgisk behandling av brudd og / eller beindeformiteter;
- Tannpleie, for å ivareta tannhelsen;
- Smertelindrende behandlinger ved svært smertefulle multiple frakturer;
- Fysioterapi, for muskelforlengelse og -forsterkning Et elastisk og tonisk muskulaturapparat lar deg forhindre fall som kan føre til forskjellige beinbrudd;
- Bruk av hjelpemidler for bevegelse, inkludert rullestoler, seler, krykker, etc.
FORDELER MED BEVEGELSE
For personer med osteogenesis imperfecta anbefaler leger konstant trening av fysisk trening og bevegelse generelt, ettersom begge disse aktivitetene bidrar til å styrke skjelett- og muskelsystemene.
Blant de anbefalte idrettsgrenene er: svømming, siden det er en "lav påvirkning fysisk aktivitet på skjelettsystemet", og gåing.
FORDELER FRA EN SUNN LIVSSTIL
Å leve et sunt liv, unngå å røyke, drikke for mye alkohol, spise for mye og dårlig osv., Har mer enn diskrete helsemessige fordeler for pasienter med osteogenesis imperfecta, ettersom det bremser sykdommens utvikling og reduserer beinets skjørhet.
SYMPTOMATISKE BEHANDLINGER I EKSPERIMENTASJONSFASEN
For tiden evaluerer leger og forskere effektiviteten av noen symptomatiske behandlinger, inkludert behandling med veksthormon og bisfosfonatbasert intravenøs og oral behandling.
For øyeblikket lover resultatene fra de nevnte undersøkelsesbehandlingene godt for hele det medisinske samfunnet.
Prognose
Osteogenesis imperfecta er en sykdom med en negativ prognose, ettersom den er uhelbredelig, kompromitterer livskvaliteten drastisk og i noen tilfeller forårsaker for tidlig død av det berørte individet.
Imidlertid bør det bemerkes at også mange mennesker med en mild form for osteogenese imperfecta, takket være moderne symptomatiske behandlinger, kan leve et hyggelig og tilfredsstillende liv.
Forebygging
Dessverre er det foreløpig ingen forebyggende tiltak mot osteogenesis imperfecta.