Definisjon av thalassemi
Thalassemi er en genetisk overført blodforstyrrelse der kroppen syntetiserer en unormal form for hemoglobin.
Som de fleste vet, er hemoglobin et protein som finnes i røde blodlegemer, avgjørende for transport av oksygen i blodet. Hos personer som lider av thalassemi, forårsaker den muterte formen for hemoglobin gradvis, men ubønnhørlig ødeleggelse av røde blodlegemer opp til anemi.
Fra medisinsk statistikk er det klart at thalassemi hovedsakelig rammer innbyggerne i land i Midtøsten, afrikanske land og alle som befolker sumpete steder (ikke overraskende kalles thalassemi også Middelhavsanemi).
Klassifisering og årsaker
I følge den defekte proteinunderenheten (som utgjør hemoglobin), skilles to former for thalassemi; før vi fortsetter med analysen, la oss ta et skritt tilbake for å klargjøre noen veldig viktige begreper.
Hemoglobin er bæreren par excellence, som brukes til å transportere oksygen i blodet; den består av to proteiner, kjent som alfa-globulin og beta-globulin.
Thalassemi oppstår når ett eller flere gener som styrer produksjonen av ett eller begge disse proteinene er defekte (mutert).
Thalassemi er forårsaket av en DNA -mutasjon av proteinene som utgjør hemoglobin: disse endringene har stor innvirkning på den fysiologiske syntesen av hemoglobin og, ved å ødelegge erytrocytter, fører det til anemi.
Klassifiseringen av thalassemi må gjøres på grunnlag av to viktige faktorer:
- Antall muterte gener arvet fra foreldre
- Type protein involvert (alfa- eller beta -hemoglobin)
Alpha Thalassemia
I "alfa" -formen for thalassemi - der de fire "alfa" kuleformede underenhetene til hemoglobin (ved kromosom 16) kan muteres - er ett eller flere defekte gener involvert; hver kuleformede underenhet er tydelig kodet fra et gen, derfor er gener som kan være involvert er 4.

Det generelle symptombildet blir mer alvorlig når tre eller fire gener er involvert: i det første tilfellet snakker vi om "hemoglobin H sykdom"(Med moderate eller alvorlige symptomer). Når alle fire genene er involvert, kalles sykdommen alfa-thalassemi major: i lignende situasjoner dør den nyfødte kort tid før fødselen eller like etter.
Beta Thalassemi
Beta -formen for thalassemi, som kan gjettes, oppstår når genene som er involvert i betakjedens sammensetning er mutert (på nivået av kromosom 11): i dette tilfellet kan bare to gener påvirkes. Hvis bare ett gen blir endret, blir det referert til som beta-thalassemi mindre, der pasienten ikke klager over relevante symptomer. På samme måte som alfa -varianten, resulterer involvering av begge gener som utgjør betakjedene til hemoglobin i en beta-thalassemi major (eller Cooley's anemi), som gjenspeiler alvorlige og alvorlige symptomer; i dette tilfellet begynner imidlertid symptomene vanligvis etter et par år fra fødselen.
Se videoen
- Se videoen på youtube
Symptomer
For ytterligere informasjon: Thalassemia Symptomer
Thalassemi er en veldig alvorlig arvelig sykdom, så mye at noen av dens varianter, for eksempel alfa-thalassemi major, kan få barnet til å dø under fødselen eller like etter fødselen. Spedbarn med beta-thalassemi major kan imidlertid overleve og utvikle seg de første symptomene innen et par år etter fødselen (alvorlig anemi).
Hvis bare ett gen blir endret, både i alfa- og beta -formen av thalassemi, klager pasientene ikke over nevneverdige symptomer; bare gjennom mikroskopanalysen av en blodprøve tatt fra pasienten er det en abnormitet i form og struktur på erytrocytter, mye mindre enn normen.
I tillegg til anemi, kan pasienter med thalassemi oppleve ett eller flere av følgende symptomer: tretthet, humørsvingninger (irritabilitet), vekstsvikt, beindeformiteter i ansiktet, gulsott, kortpustethet og mørk urin.
I alvorlige tilfeller kan det symptomatologiske bildet av en pasient som lider av thalassemi degenerere, til det vil si å skape reelle beindeformiteter, spesielt i ansiktet og hodeskallen; thalassemi kan fremme en "unormal ekspansjon av benmargen, både ved å gjøre beinmassen skjør og ved å øke risikoen for beinbrudd enormt.
Blant komplikasjonene av thalassemi, den mulige akkumuleringen av jern (hemokromatose), et uttrykk for både sykdommen selv og for de tilbakevendende blodoverføringene som pasienten trenger, bør også nevnes.
Thalassemi forårsaker ofte splenomegali, det vil si en overdreven volumetrisk økning i milten: ofte krever denne patologiske kliniske tilstanden miltomi, kirurgisk fjerning av organet. Som vi vet er milten et viktig organ som brukes til syntese av blodceller og antistoffer, i tillegg til infeksjonskontroll: fjerning av den favoriserer tydelig en reduksjon i forsvarsfunksjonen mot bakterielle og virale fornærmelser, noe som gjør motivet mer følsomt for infeksjoner. Det bør imidlertid påpekes at thalassemi i seg selv også øker risikoen for å smitte med infeksjoner: ved milt -utskæring i forbindelse med thalassemi øker sjansen for infeksjon overdrevet.

Diagnose
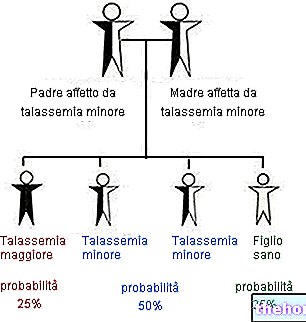
Hvis far og / eller mor er rammet av thalassemi, er sannsynligheten for å overføre sykdommen til avkommet svært høy. Vi har analysert at ikke alle former for thalassemi begynner med en presis symptomatologi helt fra fødselen: i lignende situasjoner, ved mistanke om thalassemi, er det mulig å utsette pasienten for en rekke spesifikke tester og undersøkelser, rettet mot diagnostisk vurdering ( for eksempel bestemmelse av hemoglobin A2, som er forhøyet hos friske individer som bærer beta-thalassemiske gener).
Blant de fysiske undersøkelsene kan medisinsk palpasjon av milten noen ganger konstatere en thalassemi: splenomegali, som tidligere nevnt, utgjør et første alarmsignal for middelhavsanemi. Blodprøver er mer spesifikke og presise: i en blodprøve fra en thalassemi ser de røde blodcellene små ut og har en unormal form når de sees under et mikroskop. Videre avslører en nøye blodtelling av en pasient som lider av thalassemi alvorlig anemi: denne testen er nyttig for jerntallet i blodet, for å utføre DNA -analyse for diagnostisk vurdering av sykdommen og for å evaluere mulig mutasjon av "hemoglobin" .
På den annen side avslører elektroforese av hemoglobiner den unormale formen på oksygenbærende proteiner.
Noen varianter av thalassemi kan ikke diagnostiseres med elektroforese: i dette tilfellet vil pasienten bli utsatt for en "mutasjonsanalyse" -test, som er nyttig for å oppdage og fastslå thalassemi.
Medisiner og behandlinger
Se også: Medisiner for behandling av thalassemi
Å være en genetisk overført sykdom, er det forståelig at det - for øyeblikket - ikke er noe stoff som kan reversere sykdommen; Imidlertid er det mulig å kontrollere symptomene og forbedre pasientens livskvalitet. Valget av en behandling i stedet for en annen avhenger av typen thalassemi og alvorlighetsgraden av symptomene.
I den milde varianten av thalassemi (der for eksempel bare ett gen er endret) er det ikke nødvendig med medisiner, siden pasienten ikke klager over symptomer. Under slike omstendigheter er det fortsatt tilrådelig å utføre de nødvendige kontrollene regelmessig; Noen ganger er sporadiske blodoverføringer noen ganger nyttige (spesielt ved operasjoner og fødsler).
For moderate eller alvorlige symptomatiske former er behandlingsmetoden annerledes og kan kreve hyppige blodoverføringer eller i alvorlige tilfeller stamcelletransplantasjon.
- Blodtransfusjon: denne terapeutiske tilnærmingen kan også skape alvorlige komplikasjoner, siden hyppige transfusjoner kan favorisere en patologisk opphopning av jern i blodet (hemokromatose), som krever spesiell behandling for å eliminere jernlagring, kjent som terapikelator (med medisiner som Deferasirox og Deferiprone).For mer informasjon: les artikkelen om legemidler for behandling av hemokromatose.
- Benmargstransplantasjon: forbeholdt de mest alvorlige tilfellene der thalassemi skaper alvorlige dysfunksjoner i kroppen.






.jpg)





---pan-di-spagna-per-rotolo-dolce.jpg)